ТОР 5 статей:
Методические подходы к анализу финансового состояния предприятия
Проблема периодизации русской литературы ХХ века. Краткая характеристика второй половины ХХ века
Характеристика шлифовальных кругов и ее маркировка
Служебные части речи. Предлог. Союз. Частицы
КАТЕГОРИИ:
- Археология
- Архитектура
- Астрономия
- Аудит
- Биология
- Ботаника
- Бухгалтерский учёт
- Войное дело
- Генетика
- География
- Геология
- Дизайн
- Искусство
- История
- Кино
- Кулинария
- Культура
- Литература
- Математика
- Медицина
- Металлургия
- Мифология
- Музыка
- Психология
- Религия
- Спорт
- Строительство
- Техника
- Транспорт
- Туризм
- Усадьба
- Физика
- Фотография
- Химия
- Экология
- Электричество
- Электроника
- Энергетика
Основы химической термодинамики. Термохимия
Химическая термодинамика - один из разделов физической химии. Физическая химия - наука, которая изучает химические явления на основе физических принципов и законов (физика химических явлений). Она является теоретической основой всей химической науки и технологии химических производств, различных технологических процессов, которые применяются в нехимических отраслях промышленности.
Физическая химия тесно связана с физикой. Она изучает и устанавливает количественные взаимосвязи между химическими процессами и физическими параметрами системы.
Слово "термодинамика" происходит от греческих слов "термос" - тепло и "динамос" - сила, мощь.
Термодинамика изучает законы превращения энергии из одной формы в другую в различных процессах. Выделяют общую или физическую термодинамику (изучает общие вопросы превращения энергии);техническую термодинамику (изучает взаимопревращение между теплотой и механической работой) ихимическую термодинамику (изучает превращение различных форм энергии в ходе химической реакции и при фазовых переходах, а также способность химических систем выполнять полезную работу).
Химическая термодинамика используется для решения таких задач, как
1) предсказание о возможности протекания химической реакции;
2) о направленности химической реакции;
3) о характере химического процесса.
Основным объектом исследования термодинамики является система. Это понятие означает ту часть материального мира, которая является предметом наблюдения или исследования. Это тело или группа тел, выделенных мысленно из материального мира (газ в баллоне, образец вещества, тепловая машина и т.д.). Остальная часть материального мира - за пределами условно выделенной из него системы - называется окружением или окружающей средой. Между средой и системой возможен обмен веществом и энергией.
Термодинамическая система может быть изолированной, замкнутой или открытой. Изолированная система - это система, которая совершенно не взаимодействует с окружающей средой (обмен веществом и энергией отсутствует). В замкнутой системе невозможен обмен веществом с окружающей средой (присутствует только обмен энергией). Системы, свободные от этих ограничений, называются открытыми (присутствует обмен и веществом, и энергией).
Состояние системы можно охарактеризовать некоторыми величинами, которые называютсятермодинамическими параметрами состояния. К ним относятся температура (Т), объем (v), давление (р), концентрация (с).
Характерным для термодинамики является то, что в ней рассматриваются главным образом равновесные системы. Термодинамическая система называется равновесной, если в любой точке системы значения параметров состояния одинаковы и не изменяются самопроизвольно во времени. Параметры системы, находящейся в равновесии, являются взаимозависимыми, и с изменением одного из них происходит изменение других. Количественно эта взаимосвязь может быть выражена в виде функциональной зависимости термодинамических параметров и называется уравнением состояния
F (p, v, T) = 0.
В зависимости от характера состояния системы функция может быть более или менее сложной. Например, для n молей идеального газа эта функция является наиболее простой:
Pv = nrt
И называется уравнением Менделеева - Клапейрона.
Любой параметр состояния системы является функцией остальных ее параметров. Например, Т = f (p, v). Такие функции в термодинамике называются функциями состояния. Значение этой функции зависит только от параметров состояния и не зависит от пути перехода системы в состояние равновесия.
Термодинамическая система может переходить из одного состояния в другое в результате протекания термодинамического процесса. Термодинамическим процессом называется совокупность последовательных состояний, через которые проходит система при взаимодействии ее с внешней средой. При этом ее параметры претерпевают изменения.
Обмен энергией между системой и окружающей средой может происходить двумя путями:
1) передача теплоты - способ передачи энергии, вызываемый разностью температур между системой и ее окружающей средой или между двумя разными системами. Такой способ передачи энергии осуществляется за счет хаотичного, беспорядочного движения молекул. Количество энергии, передаваемой таким образом, обозначается Q. Количество переданной теплоты пропорционально массе (m) системы и изменению температуры ΔТ, вызванному этой передачей энергии:
Q = m · ΔТ,
Где ΔТ = Т2 -Т1.
Если точно известно, из какого вещества состоит система, и это вещество можно охарактеризовать его удельной теплоемкостью с, то уравнение примет вид:
Q = m · c · ΔТ, [Дж].
Удельная теплоемкость (суд.) Вещества - это энергия, необходимая для повышения температуры 1 г (кг) данного вещества на 1 К, [Дж/г·К].
Молярная теплоемкость (сm) - это энергия, необходимая для повышения температуры 1 моля данного вещества на 1 К, [Дж/моль·К];
2) выполнение работы. Эта форма передачи энергии от одной системы к другой (или к окружающей среде) за счет упорядоченного, целенаправленного движения молекул. Система выполняет работу, если действует с некоторой силой, направленной на преодоление сопротивления. Например, чаще всего в химии рассматривают работу, выполняемую системой при расширении. Если действующей на систему силой является давление, то работа определяется уравнением:
A = - p Δv, [Дж],
Где Δv = v2 - v1. Знак "-" соответствует тому, что работа выполняется системой, а следовательно, система теряет энергию.
Работа и теплота не являются функциями состояния, так как их величина зависит от пути перехода системы из одного состояния в другое. Следует также отметить, что теплота и работа сами по себе не содержатся в системе. Содержится энергия в виде различных форм движения, которые в момент передачи от одной системы к другой могут стать теплотой или работой.
Кроме p, v, T к функциям состояния системы относится энергия. Это мера способности системы совершать работу. Энергия может существовать в разнообразных формах. Например, химическая, электрическая, механическая, ядерная, солнечная. Химическая энергия относится к химическим системам, солнечная - к энергии Солнца. Механическую форму можно подразделить на кинетическую (энергию, связанную с движением тела) и потенциальную (энергию, запасенную телом).
Сумма кинетической и потенциальной энергии всех частиц в системе называется внутренней энергией(U) системы.
Кинетическая энергия обусловлена движениями частиц, а потенциальная энергия обусловлена электростатическими силами притяжения между частицами и внутри частиц.
Внутренняя энергия системы является функцией состояния системы. Абсолютное значение внутренней энергии системы не поддается экспериментальному определению, поэтому в термодинамике рассматривают только изменение внутренней энергии системы:
ΔU = U2 - U1
Где U2 - внутренняя энергия системы в конечном состоянии,
U1 - внутренняя энергия системы в начальном состоянии.
ΔU имеет отрицательное значение в том случае, когда система теряет энергию, т.е. Когда энергия передается от системы к ее окружению.
Термодинамика основывается на двух основных законах, называемых первым и вторым началами. Они представляют собой аксиомы, установленные на основе множества экспериментальных фактов и опыта. Оба закона имеют несколько формулировок.
1 закон термодинамики представляет собой одну из формулировок закона сохранения энергии применительно к термодинамическим процессам. Закон сохранения энергии утверждает, что энергия не создается и не уничтожается, а только лишь превращается из одной формы в другую. 1 закон термодинамики можно сформулировать так: "Невозможно создать вечный двигатель первого рода, т.е. Машину, которая совершала бы работу, не затрачивая энергию". Вторая формулировка повторяет математическую формулу: "Изменение внутренней энергии системы (ΔU) равно сумме между количеством теплоты (Q) и количеством работы (А)"
ΔU = Q + A, [Дж].
Если работа имеет положительное значение, система приобретает энергию. Это означает, что работа выполняется над системой. Если работа выполняется самой системой, то система теряет энергию, и работа имеет отрицательное значение:
ΔU = Q - A, [Дж].
Рассмотрим некоторые закономерности, вытекающие из 1 закона или так называемые частные случаи 1 закона.
1) Изохорный процесс (V = const).
А = 0, т.к. Не происходит изменения объема.
ΔU = Qv.
Qv = n · cv · (T2 - T1).

2) Изотермический процесс (Т = const).
Внутренняя энергия системы не меняется. Вся сообщаемая теплота расходуется на совершение работы А по расширению системы.
ΔU = 0.
Qт = А.
А = nrt · 2,3 lg  (если изменяется объем).
(если изменяется объем).
А = nrt · 2,3 lg  (если изменяется давление).
(если изменяется давление).
3) Адиабатный процесс (Q = 0).
Теплообмен с окружающей средой отсутствует. Система может совершать работу только за счет убыли внутренней энергии. А = - ΔU.
A = n c (T1 - T2).
4) Изобарный процесс (р = const). ΔU = Qp - A
Qp = ΔU + A = ΔH.
ΔH - функция состояния системы, называемая энтальпией.
Таким образом, для изобарного процесса теплота его равна изменению энтальпии системы.
А = p Δv.
Qp = n cp (T2 - T1)

Итак, теплота, поглощаемая системой при постоянном давлении, равна изменению энтальпии системы ΔH = Qp.
Изменение энтальпии можно представить в виде
ΔH = Н2 - Н1,
Где Н1 - энтальпия реагентов, Н2 - энтальпия продуктов. Эту величину еще называют теплотой реакции. В зависимости от значения ΔH реакция может быть эндотермической и экзотермической.
Каждый конкретный процесс характеризуется стандартным изменением молярной энтальпии δhm° (298 К). Это изменение энтальпии при образовании 1 моль данного вещества из входящих в него элементов в стандартных условиях (Т = 298 К, р = 1 атм.).
Например, 2Н2 + О2 = 2Н2О, δhm° = - 571,6 кдж/моль.
Количество выделенной или поглощенной теплоты называется тепловым эффектом химической реакции. Уравнение химической реакции, в котором указан тепловой эффект, называется термохимическим уравнением.
Энтальпии многих реакций не поддаются экспериментальному определению по той причине, что эти реакции невозможно провести в лабораторных условиях. Они могут быть вычислены по известным энтальпиям других реакций с помощью закона Гесса. В 1840 г. Русский ученый Г. Гесс опытным путем установил факт независимости теплового эффекта химического процесса от пути его протекания. Этот закон является основным законом термохимии и гласит:
"Тепловой эффект процесса не зависит от пути перехода, а зависит только от вида и состояния исходных веществ и конечных продуктов".
Закон Гесса справедлив только для условий постоянства объема или давления:
Qv = ΔU
Qp = ΔU + A.
Рассмотрим закон Гесса на примере сгорания графита до углекислого газа. Реакцию можно проводить по двум направлениям: 1) С + О2 = СО2, ΔH
2) С + ½ О2 = СО, ΔH1
СО + ½ О2 = СО2, ΔH2.
Очевидно, что ΔH = ΔH1 + ΔH2.
Таким образом, независимо от того, сгорает ли графит сначала в СО и затем в СО2 или сразу в СО2, тепловой эффект будет одним и тем же. Из уравнения следует, что ΔH1= ΔH - ΔH2, т.е. Можно вычислить теплоту промежуточной реакции, если известны теплоты других реакций, комбинированием которых она может быть представлена. Из закона Гесса вытекают два следствия:
1) Тепловой эффект реакции равен сумме теплот образования (δhобр.) Продуктов реакции за вычетом суммы теплот образования исходных веществ с учетом стехиометрических коэффициентов.
Δhх.р. = ∑ (δhобр.)Прод. - ∑ (δhобр.)Исх.
2) Тепловой эффект реакции равен сумме теплот сгорания (δhсгор.) Исходных веществ за вычетом суммы теплот сгорания продуктов реакции с учетом стехиометрических коэффициентов.
Δhх.р. = ∑ (δhсгор.)Исх. - ∑ (δhсгор.)Прод.
Под теплотой образования δhобр. Понимают тепловой эффект образования 1 моль вещества из простых веществ, устойчивых при стандартных условиях.
Под теплотой сгорания δhсгор. Понимают тепловой эффект сгорания 1 моль вещества до СО2 и Н2О.
Стандартная молярная энтальпия образования δhm° всякого свободного элемента равна 0.
I закон термодинамики утверждает, что, хотя между системой и ее окружением возможна передача энергии, энергия никогда не создается и не исчезает. Одно время полагали, что все химические реакции являются экзотермическими, т.е. Химическая реакция может осуществляться только в том случае, если система теряет энергию. Однако в настоящее время известны многие химические и физические превращения, которые являются эндотермическими. Следовательно, по одному лишь изменению энергии или энтальпии еще нельзя предсказать, будет самопроизвольно осуществляться реакция или нет. Чтобы предсказать, возможно ли самопроизвольное протекание реакции, необходимо ввести еще одну термодинамическую функцию состояния, называемую энтропией S. Энтропию можно охарактеризовать как меру хаотичности, беспорядка в системе. Например, частицы газа в гораздо большей мере не упорядочены, чем частицы твердого вещества. Следовательно, энтропия газов, как правило, намного больше, чем энтропия твердых веществ.
Так как энтропия является функцией состояния системы, ее величина может вполне определяться параметрами состояния системы:
S = f (p, T, v).
Существование энтропии определяет II закон термодинамики. Он также имеет несколько формулировок:
1. "Теплота сама собой не может переходить от холодного тела к горячему" (Клаузиус);
2. "Невозможен процесс, единственным результатом которого было бы превращение теплоты в работу" (Томсон);
3. "Невозможно создать вечный двигатель второго рода, т.е. Машину, все действие которой сводится к производству работы и охлаждению теплоисточника" (Оствальд).
Поясним последнюю формулировку. Невозможно построить такую машину, так как любая тепловая машина должна иметь холодильник с более низкой температурой, чем теплоисточник. Поэтому часть теплоты от теплоисточника (котла) совершает работу, а другая часть переходит к холодильнику.

Схема перехода теплоты в работу
II закон термодинамики является одним из наиболее общих положений всей науки в целом. Главная мысль его заключается в том, что в любой изолированной системе с течением времени происходит постоянное возрастание степени беспорядка, т.е. Энтропии. Следовательно, для любых самопроизвольных процессов
ΔS ≥ 0.
Знак ">" - для необратимых процессов, знак "=" - для обратимых процессов.
Для обратимых процессов ΔS = Q/T, [ Дж/К · моль].
Для необратимых процессов ΔS > Q/T, [ Дж/К · моль].
Термодинамика изучает макросистемы. Любое макросостояние системы может быть определено множеством микросостояний.
Число микросостояний, с помощью которых может быть осуществлено данное макросостояние, называетсятермодинамической вероятностью ω. Между энтропией и термодинамической вероятностью существует функциональная связь, выраженная формулой Больцмана:
S = k · lnω,
Где k - константа Больцмана, равная 1,38·10-16.
Все процессы, связанные с увеличением упорядоченности (уменьшения числа микросостояний) (отвердевание, кристаллизация, сжатие, конденсация), сопровождаются уменьшением энтропии.
II закон термодинамики имеет ясный физический смысл только тогда, когда его применяют к любой ограниченной системе.
Функции системы, которые связаны с работой и говорящие о направлении процесса, называются термодинамическими потенциалами.
Критерием для суждения о направлении процессов в изолированных системах может служить изменение энтропии ΔS. Однако на практике большинство процессов протекает в неизолированных системах и связано с теплообменом и изменением объема. Поэтому для неизолированных систем необходимо иметь другие критерии.
Для процессов, протекающих при постоянных Т и V, применяется изохорно-изотермический потенциал (свободная энергия Гельмгольца F):
ΔF = U - T · S.
Для процессов, протекающих при постоянных Т и р, критерием, определяющим направление течения процесса, будет являться другая термодинамическая функция - изобарно-изотермический потенциал (свободная энергия Гиббса G):
ΔG = H - T · S.
Критерии обратимости и необратимости процессов, при которых совершается только работа расширения
| Условия, при которых протекает процесс | Обратимый равновесный процесс | Необратимый процесс |
| U, S - const | ΔU = 0 | ΔU < 0 |
| V, U - const | ΔS = 0 | ΔS > 0 |
| T, V - const | ΔF = 0 | ΔF < 0 |
| T, p - const | ΔG = 0 | ΔG < 0 |
54.55.56.
О принципиальной возможности протекания химических реакций судят по величине изменения свободной энергии системы 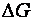 . Однако этого недостаточно, чтобы предсказать реальную возможность химического процесса, определить скорость реакции, ее механизм, а также управлять процессом.
. Однако этого недостаточно, чтобы предсказать реальную возможность химического процесса, определить скорость реакции, ее механизм, а также управлять процессом.
Например, термодинамическая вероятность окисления водорода до газообразной воды
Н2(Г) + 1/2О2(Г) = Н2О(Г), 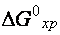 = –228,3 кдж/моль
= –228,3 кдж/моль
Значительно выше, чем вероятность реакции окисления оксида азота (II)
NO(Г) + 1/2О2(Г) = NO2(Г), = –35,1 кдж/моль.
В то же время первая реакция при комнатной температуре практически не протекает, и смесь водорода с воздухом может храниться в этих условиях длительное время, вторая же реакция при тех же условиях протекает мгновенно. Таким образом, для полного описания химического процесса необходимо знать закономерность протекания его во времени. Скорость и механизм химических реакций изучает химическая кинетика.
3.2.1. Скорость химических реакций
Различают гомогенные и гетерогенные химические реакции.
Гомогенные – это реакции, протекающие между веществами, находящимися в однородной среде, т. Е. Между ними нет границы раздела (газовая фаза, жидкая фаза). Реакции протекают равномерно по всему объему системы:
2naoh(Ж) + H2SO(Ж) = Na2SO4(Ж) + 2Н2О(Ж).
Гетерогенные – это реакции в неоднородной среде, протекающие между веществами, находящимися в разных фазовых состояниях (твердое и жидкое вещество, газообразное и твердое вещество и пр.). Такие реакции протекают только на поверхности раздела фаз, образующих эту систему:
Caco3(Т) + 2hcl(Ж) = cacl2(Ж) + CO2(Г) + Н2О(Ж).
Под скоростью химической реакции  понимается изменение количества вещества (моль) в единицу времени в единице реакционного пространства. Для гомогенных реакций реакционное пространство – объем, , моль/л∙ед. Времени, гетерогенных – площадь поверхности раздела фаз, , моль/м2∙ед. Времени.
понимается изменение количества вещества (моль) в единицу времени в единице реакционного пространства. Для гомогенных реакций реакционное пространство – объем, , моль/л∙ед. Времени, гетерогенных – площадь поверхности раздела фаз, , моль/м2∙ед. Времени.
Так как отношение количества вещества к единице объема называется концентрацией вещества (С, моль/л), то скорость гомогенного процесса равна изменению концентрации чаще исходных веществ во времени:
 (36)
(36)
Где  – средняя скорость реакции, моль/л∙ед. Времени;
– средняя скорость реакции, моль/л∙ед. Времени;  – изменение концентрации исходных веществ, моль/л;
– изменение концентрации исходных веществ, моль/л; 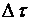 – время протекания процесса, с, мин и пр.
– время протекания процесса, с, мин и пр.
Скорость реакций зависит от ряда факторов: природы реагирующих веществ, их концентрации, температуры и давления процесса, наличия в системе катализатора, а в случае гетерогенных реакций – от состояния поверхности раздела фаз.
3.2.2. Зависимость скорости реакции от концентрации реагентов
Чтобы произошла химическая реакция необходимо столкновение молекул реагирующих веществ. Число таких столкновений растет с увеличением числа молекул в единице объема, т. Е. С возрастанием концентрации реагентов. Соответственно, с повышением концентрации реагирующих веществ увеличивается скорость реакции.
Зависимость скорости реакции от концентрации реагирующих веществ определяетсязаконом действия масс, который был открыт опытным путем в 1867 г. К. Гульдбергом и П. Вааге (Норвегия).
Скорость химической реакции, протекающей при постоянной температуре в гомогенной среде, прямо пропорциональна произведению концентраций реагирующих веществ, возведенных в степени их стехиометрических коэффициентов, называемых порядками реакции по реагентам.
Рассмотрим гомогенный процесс окисления оксида азота (II):
2NO(Г) + O2(Г) = 2NO2(Г).
Обозначим концентрации реагирующих веществ:  . Математическое выражение закона действия масс для процесса примет вид:
. Математическое выражение закона действия масс для процесса примет вид:
 (37)
(37)
Где 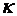 – константа скорости реакции.
– константа скорости реакции.
Константа скорости реакции зависит от химической природы реагирующих веществ, температуры процесса, наличия в системе катализатора, но не зависит от концентрации реагирующих веществ.
Уравнение (37) называется кинетическим уравнением химической реакции.
Сумма показателей степеней при концентрациях веществ в кинетическом уравнении (n) определяет молекулярность или порядок реакции. Для уравнения (37) порядок равен: n = 2 + 1 = 3. Для одностадийных процессов n ≤ 3. При большем порядке реакции протекают в несколько стадий.
В случае гетерогенных реакций в математическое выражение закона действия масс входят концентрации только тех веществ, которые находятся в газовой фазе или в растворе. Концентрации веществ, находящихся в твердой фазе, представляют собой постоянную величину и входят в константу скорости реакции ( ). Например, для реакции горения угля:
С(Т) + О2(Г) = СО2(Г), кинетическое уравнение имеет вид:
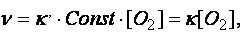 (38)
(38)
Где  .
.
Зависимость скорости реакции от природы реагирующих веществ и температуры
Химическая реакция (разрыв связей между атомами в молекулах исходных веществ и образование новых связей между атомами в молекулах продуктов реакции, (см. Рис. 32, подразд. 3.1) протекает при непосредственном столкновении молекул исходных веществ, причем последние должны обладать при этом достаточным запасом кинетической энергии, иначе их столкновение будет неэффективным.
Избыточная кинетическая энергия, которой должны обладать молекулы исходных веществ для того, чтобы их столкновение могло привести к образованию нового вещества, называется энергией активации данного процесса  , кдж/моль. Молекулы, обладающие такой энергией, называются активными.
, кдж/моль. Молекулы, обладающие такой энергией, называются активными.
Энергия активации – это фактор, посредством которого природа реагирующих веществ влияет на скорость реакции. Значения для химических реакций лежат в пределах 40….400 кдж/моль. Установлено, если:
< 40 кдж/моль, скорость очень велика, реакции протекают практически мгновенно (ионные реакции в растворах);
> 120 кдж/моль, скорость очень мала, реакции при обычных условиях визуально не видны (синтез аммиака);
≈ 40….120 кдж/моль, скорость характерна для большинства химических реакций, её можно замерить.
В химических реакциях выделяется три последовательно сменяющихся энергетических состояния, примером может служить схема реакции синтеза иодистого водорода HJ:

Активированный комплекс возникает в качестве промежуточного состояния в ходе как прямой, так и обратной реакции. В состоянии активированного комплекса старые связи еще не разрушены, но уже ослаблены, новые связи наметились, но еще не образовались. Время существования его не велико (10–14….10–11 с). Энергетически он отличается от исходных веществ и продуктов реакции (рис. 35).



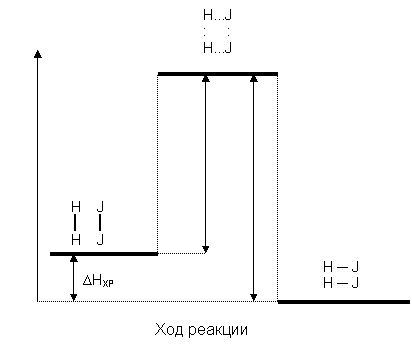
Рис. 35. Энергетическая схема реакции cинтеза HJ
Энергия, необходимая для перехода вещества в состояние активированного комплекса, называется энергией активации прямого или обратного процессов, при этом прямой реакции ≠ Еа обратной реакции.
В 1889 г. Шведский ученый С. Аррениус вывел уравнение, которое носит его имя:
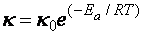 , (39)
, (39)
Где – константа скорости реакции; 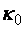 – предэкспоненциальный множитель (отражает частоту столкновений и ориентацию реагирующих частиц);
– предэкспоненциальный множитель (отражает частоту столкновений и ориентацию реагирующих частиц);  – основание натурального логарифма; – энергия активации процесса;
– основание натурального логарифма; – энергия активации процесса;  – универсальная газовая постоянная (8,314 Дж/моль∙К);
– универсальная газовая постоянная (8,314 Дж/моль∙К); 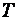 – абсолютная температура процесса, К.
– абсолютная температура процесса, К.
Из уравнения Аррениуса следует, что константа скорости реакции, а, следовательно, и скорость реакции, уменьшается с ростом энергии активации. Повышение же температуры процесса приводит к увеличению константы и скорости реакции.
Зависимость скорости реакции (константы скорости) от температуры определяется правилом Я. Вант-Гоффа (Голландия, 1884 г.).
При повышении температуры на 10 градусов скорость большинства реакций увеличивается в 2–4 раза:
 , (40)
, (40)
Где  и
и  – скорости реакций при температурах
– скорости реакций при температурах 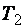 и
и  ;
;  – температурный коэффициент скорости реакции.
– температурный коэффициент скорости реакции.
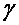 – это число, показывающее, во сколько раз возрастает скорость данной реакции при повышении температуры системы на 10 градусов.
– это число, показывающее, во сколько раз возрастает скорость данной реакции при повышении температуры системы на 10 градусов.
С увеличением температуры растет число молекул, кинетическая энергия которых равна или выше энергии активации , следовательно, растет доля молекул, способных к активным столкновениям с образованием активированного комплекса, т. Е. Происходит ускорение реакции.
3.2.4. Механизмы химических реакций
Под механизмом химических реакций понимается последовательность протекания простейших стадий реакции. Классификация химических реакций по механизмам протекания представлена на рис. 36.

Рис. 36. Классификация химических реакций по механизму протекания
Число молекул реагентов, принимающих участие в элементарной стадии реакции, называется молекулярностью реакции. Одностадийные реакции с молекулярностью более трех неизвестны.
Простые (одностадийные) реакции протекают через образование активированного комплекса, порядок реакции совпадает с молекулярностью, а кинетическое уравнение процесса – с законом действующих масс. К реакциям подобного типа относятся реакции:
– диссоциации 2HJ → H2 + J2,  ;
;
– соединения NO + O3 = NO2 + O2, 
К сложным реакциям относятся реакции, протекающие последовательно или параллельно через несколько стадий. Скорость реакции будет определять самая медленная (лимитирующая) стадия процесса. Например, реакция разложения N2O5, 2N2O5 = 4NO2 + O2 протекает через несколько стадий:
N2O5 ↔ NO2+ NO3 (быстрая стадия);
NO2 + NO3 → NO2 + NO + O2 (медленная стадия);
NO + NO3 ↔ 2NO2 (быстрая стадия).
Лимитирующей является вторая (бимолекулярная) стадия процесса:  , порядок реакции в данном случае (n = 1 + 1 = 2) совпадает с молекулярностью суммарной реакции разложения N2O5, однако такое равенство не всегда имеет место.
, порядок реакции в данном случае (n = 1 + 1 = 2) совпадает с молекулярностью суммарной реакции разложения N2O5, однако такое равенство не всегда имеет место.
В последнее время все больше внимания уделяется периодическим процессам (колебательным реакциям). Такие периодические процессы характеризуются колебаниями концентраций некоторых промежуточных соединений и, соответственно, скоростей этих стадий процесса. Реакции были открыты в середине прошлого века Б.П. Белоусовым (Россия).
Примером подобных процессов является реакция окисления малоновой кислоты СН3(СООН)2 избытком перекиси водорода Н2О2 в присутствии иодат ионов JO3– и ионов марганца Mn2+, играющих роль катализатора, а также индикатора – крахмала. На рис. 37 представлена схема протекания этого процесса.

|
Рис. 37. Схема колебательной реакции окисления малоновой кислоты
В ходе реакции окраска раствора периодически меняется: желтая, темно-синяя, бесцветная, – затем цикл повторяется. Колебания прекращаются после полного окисления малоновой кислоты. Изменение окраски связано с периодическим изменением концентрации: JO3–, J2, J2∙крахмал. Период колебаний зависит от концентрации веществ и температуры процесса.
Механизм протекания колебательных реакций очень сложен и объясняется на основе термодинамически необратимых процессов.
В начале прошлого века российские ученые Н.А. Шилов и Н.Н. Семенов разработали теорию цепных реакций. Такие реакции встречаются часто: горение топлива в двигателях внутреннего сгорания, реакции полимеризации, реакции, протекающие в атмосфере и пр.
Цепные реакции начинаются со стадии инициирования, т. Е. Образования активных частиц (свободных радикалов), которые представляют собой осколки молекул, имеющих неспаренные электроны: Cl*, O*, HS* и др. Свободные радикалы образуются при воздействии на систему света, тепла, излучения высокой энергии, либо в ходе экзотермических процессов (горение органического топлива в двигателях внутреннего сгорания). Появление свободных радикалов называется стадией зарождения цепи. Далее идет рост цепи – радикалы взаимодействуют с молекулами, образуя продукты реакции и новые радикалы. Процесс заканчивается обрывом цепи, когда радикалы, взаимодействуя друг с другом или с молекулами, образуют другие нейтральные молекулы.
57.58.59.
Понятия о катализе
Наиболее мощным способом интенсификации химических процессов является применение катализаторов.
Катализаторы – это вещества, которые ускоряют химические процессы, но при этом не испытывают превращений в ходе реакции. Явление изменения скорости реакции под действием таких веществ называется катализом.
Как правило, катализаторы обладают селективным, т. Е. Избирательным действием, подбирая вид катализатора, можно изменить ход протекания реакции.
Например, этанол С2Н5ОН в присутствии оксидов алюминия и тория разлагается на этилен С2Н4 и воду:
С2Н5ОН 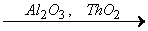 С2Н4 + Н2О,
С2Н4 + Н2О,
В присутствии никеля, железа, серебра или меди – на ацетальдегид СН3СОН и водород:
С2Н5ОН  СН3СОН + Н2.
СН3СОН + Н2.
Сущность действия катализаторов очень сложна и до конца не изучена. Предполагается, что в каталитических процессах снижается энергия активации реакции (см. Рис. 35), так как в присутствии катализатора образуются другие промежуточные активированные комплексы, которые требуют меньшей энергии образования, чем в реакциях, протекающих без катализатора. Многие молекулы исходных веществ, энергия которых была недостаточна для активных столкновений в обычных реакциях, в присутствии катализатора становятся активными. Следовательно, 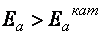 . Катализаторы увеличивают скорость реакции , но не влияют на термодинамику процесса, т. Е. Не изменяют величины
. Катализаторы увеличивают скорость реакции , но не влияют на термодинамику процесса, т. Е. Не изменяют величины  и
и  . На активность катализатора влияют промоторы – вещества, при добавлении в небольших количествах которых, эффективность действия катализатора повышается, икаталитические яды – вещества, снижающие каталитическую активность.
. На активность катализатора влияют промоторы – вещества, при добавлении в небольших количествах которых, эффективность действия катализатора повышается, икаталитические яды – вещества, снижающие каталитическую активность.
Различают гомогенный и гетерогенный катализ.
При гомогенном катализе катализатор и реагирующие вещества находятся в одном фазовом состоянии (газ, жидкость).
Пример, промышленное получение серной кислоты.
Реакция окисления: 2H2SO3 + O2 = 2H2SO4 – протекает медленно, она заменяется на быстрые каталитические реакции, где катализатором является NO:
O2 + 2NO = 2NO2, NO2 + H2SO3 = H2SO4 + NO.
Таким образом, гомогенные каталитические реакции протекают через образование промежуточных соединений, в которых участвует катализатор.
Многие природные и физиологические процессы, катализируемые ферментами, протекают по механизму гомогенного катализа: расщепление белков, дегидратация СО2 из крови и т. Д.
При гетерогенном катализе катализатор и реагенты находятся в разных фазовых состояниях, чаще всего катализатор – твердое вещество, и реакция протекает на поверхности катализатора. Скорость такой реакции зависит от площади поверхности катализатора, поэтому последний часто наносят на вещества с развитой поверхностью – подложку (пористые угли, силикаты и пр.).
Как и в случае гомогенного катализа, при гетерогенном катализе реакция протекает через образование активных промежуточных соединений, которые представляют собойповерхностные соединения катализатора с реагирующими веществами. Проходя через ряд стадий (подвод реагентов в зону реакции, адсорбция на поверхности катализатора, собственно химическая реакция, десорбция), в которых участвуют и промежуточные соединения, реакция заканчивается образованием конечных продуктов, а катализатор – не расходуется.
Гетерогенный катализ в промышленности применяется при получении аммиака, азотной и серной кислот, водорода и пр. К наиболее распространенным катализаторам относятся: Pt,Ni, Pd, cuo, V2O5, Al2O3, sio2 и др. Применение каталитических процессов обеспечивает экономию сырья и энергии, а также решает экологические задачи. Например, катализаторы применяют для доокисления токсичных выхлопных газов в двигателях внутреннего сгорания до нетоксичных компонентов:
Cnhm + qo2  ncо2 + m/2Н2О,
ncо2 + m/2Н2О,
2NO + 2CO  N2 +2CO2 + 690 кдж (в качестве катализатора используется металлическая вата из нержавеющей стали или сплавы никеля).
N2 +2CO2 + 690 кдж (в качестве катализатора используется металлическая вата из нержавеющей стали или сплавы никеля).
3.2.6. Необратимые и обратимые реакции, химическое равновесие
Все химические реакции делятся на две группы: необратимые и обратимые.
Необратимые реакции протекают в одном направлении, до полного израсходования одного из реагирующих веществ, обязательное условие их протекания – удаление продуктов реакции из сферы реакции (в виде газа, нерастворимого соединения или слабого электролита).
Пример:
Zn + 4HNO3 = Zn(NO3)2 + 2NO2↑ + 2H2O.
Обратимые реакции протекают в двух взаимно противоположных направлениях, реакция не идет до конца, т. Е. Ни одно из реагирующих веществ не расходуется полностью, реакция идет до определенного предела, называемого химическим равновесием.
Рассмотрим обратимый процесс: H2 + J2 ↔ 2HJ.
В начальный момент времени (  ) концентрации исходных веществ равны:
) концентрации исходных веществ равны:  , а концентрация продуктов реакции отсутствовала:
, а концентрация продуктов реакции отсутствовала:  = 0. При этих условиях реакция протекает только в прямом направлении, кинетическое уравнение имеет вид:
= 0. При этих условиях реакция протекает только в прямом направлении, кинетическое уравнение имеет вид:
 . (41)
. (41)
Скорость обратной реакции в начальный момент времени равна нулю:
 . (42)
. (42)
В ходе прямой реакции концентрации водорода и иода непрерывно уменьшаются, в соответствии с этим пропорционально снижается скорость прямого процесса. Одновременно с этим нарастает концентрация продукта реакции HJ и в момент времени 
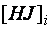 ≠ 0. Появляются условия, определяющие протекание обратной реакции, кинетическое уравнение обратного процесса имеет вид:
≠ 0. Появляются условия, определяющие протекание обратной реакции, кинетическое уравнение обратного процесса имеет вид:
 . (43)
. (43)
По мере нарастания концентрации иодоводорода HJ увеличивается скорость обратной реакции, в момент времени  , когда скорости прямой и обратной реакции выравниваются:
, когда скорости прямой и обратной реакции выравниваются:
 , (44)
, (44)
Наступает динамическое химическое равновесие системы.
Химическое равновесие – это такое состояние системы, при котором скорости прямого и обратного процессов равны, а концентрации всех веществ, участвующих в процессе, перестают изменяться и называются равновесными.
Состояние равновесия характеризует тот предел, к которому в данных условиях обратимая реакция протекает самопроизвольно,  → 0.
→ 0.
В формуле (44) выразим скорости реакций через равновесные концентрации реагирующих веществ:
 . (45)
. (45)
Перенесем константы в одну сторону уравнения, а равновесные концентрации – в другую:
 . (46)
. (46)
Отношение констант скоростей прямого и обратного процессов тоже представляет собой постоянную величину, которая называется константой равновесия данного равновесного процесса  . Для равновесного процесса, протекающего при постоянной температуре, в общем случае: ma + nb ↔ pc + qd, константа равновесия выразится уравнением:
. Для равновесного процесса, протекающего при постоянной температуре, в общем случае: ma + nb ↔ pc + qd, константа равновесия выразится уравнением:
 , (47)
, (47)
Где  – равновесные концентрации продуктов реакции;
– равновесные концентрации продуктов реакции;  – равновесные концентрации исходных веществ,
– равновесные концентрации исходных веществ,  – стехиометрические коэффициенты в уравнении реакции.
– стехиометрические коэффициенты в уравнении реакции.
Константа равновесия является количественной характеристикой равновесного процесса:
– для необратимых реакций, прошедших до конца, →  , так как концентрация продуктов реакции значительно больше концентрации исходных веществ;
, так как концентрация продуктов реакции значительно больше концентрации исходных веществ;
– при полном отсутствии химического взаимодействия,  , так как концентрация исходных веществ значительно превосходит концентрацию продуктов реакции.
, так как концентрация исходных веществ значительно превосходит концентрацию продуктов реакции.
В случае гетерогенных равновесных процессов в выражение константы равновесия входят только концентрации тех веществ, которые находятся в газовой или жидкой фазе.
Пример: CO2(Г) + С(К) ↔ 2СО(Г),
 . (48)
. (48)
Константа равновесия реакции связана с изменением энергии Гиббса процесса зависимостью:
 . (49)
. (49)
Уравнение (49) справедливо для любой температуры, но чаще применяется для стандартных условий: 25 °С (298 К). При подстановке значения газовой постоянной ( = 8,314 Дж/ моль∙К) и стандартной температуры уравнение (49) примет вид:
 . (50)
. (50)
Это уравнение позволяет, зная значение , вычислять константу равновесия и, наоборот, по экспериментально найденному значению  определять .
определять .
Смещение химического равновесия, принцип Ле Шателье
Состояние химического равновесия зависит от ряда факторов: концентрации реагирующих веществ, температуры, давления в системе. Если условия изменяются, система выходит из состояния равновесия, при этом скорости прямой и обратной реакций изменяются не пропорционально. Через некоторое время равновесие устанавливается при новых внешних условиях.
Переход из одного равновесного состояния в другое, отвечающее новым внешним условиям, называется сдвигом или смещением химического равновесия.
В 1884 г. Ле Шателье (Франция) сформулировал принцип, определяющий влияние различных факторов на равновесные системы.
Если на систему, находящуюся в равновесии, оказать какое-либо воздействие (изменить концентрацию веществ, температуру или давление), то в результате протекающих в ней процессов равновесие сместится в том направлении, что оказанное воздействие уменьшится.
60.
Зако́ны Ра́уля — общее название открытых французским химиком Ф. М. Раулем в 1887 г. Количественных закономерностей, описывающих некоторые коллигативные (зависящие от концентрации, но не от природы растворённого вещества) свойства растворов.Содержание [убрать]
Первый закон Рауля связывает давление насыщенного пара над раствором с его составом; он формулируется следующим образом:
Парциальное давление насыщенного пара компонента раствора прямо пропорционально его мольной доле в растворе, причём коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом.
 Для бинарного раствора, состоящего из компонентов А и В (компонент А считаем растворителем) удобнее использовать другую формулировку:
Для бинарного раствора, состоящего из компонентов А и В (компонент А считаем растворителем) удобнее использовать другую формулировку:
Относительное понижение парциального давления пара растворителя над раствором не зависит от природы растворённого вещества и равно его мольной доле в растворе.
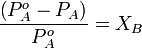 На поверхности оказывается меньше способных испаряться молекул растворителя, ведь часть места занимает растворённое вещество.
На поверхности оказывается меньше способных испаряться молекул растворителя, ведь часть места занимает растворённое вещество.
Растворы, для которых выполняется закон Рауля, называются идеальными. Идеальными при любых концентрациях являются растворы, компоненты которых очень близки по физическим и химическим свойствам (оптические изомеры, гомологи и т. П.), и образование которых не сопровождается изменением объёма и выделением либо поглощением теплоты. В этом случае силы межмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором.
Второй закон Рауля
Тот факт, что давление паров над раствором отличается от давления паров над чистым растворителем, существенно влияет на процессы кристаллизации и кипения. Из первого закона Рауля выводятся два следствия, касающиеся понижения температуры замерзания и повышения температуры кипения растворов, которые в объединённом виде известны как второй закон Рауля.
Понижение температуры кристаллизации растворов
Условием кристаллизации является равенство давления насыщенного пара растворителя над раствором давлению пара над твёрдым растворителем. Поскольку давление пара растворителя над раствором всегда ниже, чем над чистым растворителем, это равенство всегда будет достигаться при температуре более низкой, чем температура замерзания растворителя. Так, океанская вода начинает замерзать при температуре около минус 2 °C.
Разность между температурой кристаллизации растворителя T°fr и температурой начала кристаллизации раствора Tfr есть понижение температуры кристаллизации.
Понижение температуры кристаллизации бесконечно разбавленных растворов не зависит от природы растворённого вещества и прямо пропорционально моляльной концентрации раствора.
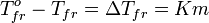
Поскольку по мере кристаллизации растворителя из раствора концентрация последнего возрастает, растворы не имеют определённой температуры замерзания и кристаллизуются в некотором интервале температур.
Повышение температуры кипения растворов
Жидкость кипит при той температуре, при которой общее давление насыщенного пара становится равным внешнему давлению. Если растворённое вещество нелетуче (то есть давлением его насыщенных паров над раствором можно пренебречь), то общее давление насыщенного пара над раствором равно парциальному давлению паров растворителя. В этом случае давление насыщенных паров над раствором при любой температуре будет меньше, чем над чистым растворителем, и равенство его внешнему давлению будет достигаться при более высокой температуре. Таким образом, температура кипения раствора нелетучего вещества Tb всегда выше, чем температура кипения чистого растворителя при том же давлении T°b.

Повышение температуры кипения бесконечно разбавленных растворов нелетучих веществ не зависит от природы растворённого вещества и прямо пропорционально моляльной концентрации раствора
Криоскопическая и эбулиоскопическая константы
Коэффициенты пропорциональности К и Е в приведённых выше уравнениях — соответственно криоскопическая и эбулиоскопическая постоянные растворителя, имеющие физический смысл понижения температуры кристаллизации и повышения температуры кипения раствора с концентрацией 1 моль/кг. Для воды они равны 1.86 и 0.52 K·моль−1·кг соответственно. Поскольку одномоляльный раствор не является бесконечно разбавленным, второй закон Рауля для него в общем случае не выполняется, и величины этих констант получают экстраполяцией зависимости из области малых концентраций до m = 1 моль/кг.
Для водных растворов в уравнениях второго закона Рауля моляльную концентрацию иногда заменяют молярной. В общем случае такая замена неправомерна, и для растворов, плотность которых отличается от 1 г/см³, может привести к существенным ошибкам.
Второй закон Рауля даёт возможность экспериментально определять молекулярные массы соединений, неспособных к диссоциации в данном растворителе; его можно использовать также для определения степени диссоциации электролитов.
Растворы электролитов
Законы Рауля не выполняются для растворов (даже бесконечно разбавленных), которые проводят электрический ток — растворов электролитов. Для учёта этих отклонений Вант-Гофф внёс в приведённые выше уравнения поправку — изотонический коэффициент i, неявно у 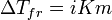

Неподчинение растворов электролитов законам Рауля и принципу Вант-Гоффа послужили отправной точкой для создания С. А. Аррениусом теории электролитической диссоциации.
Свойства растворов
Все растворы обладают некоторыми свойствами, которые практически зависят только от соотношения числа частиц компонентов раствора и не зависят от природы частиц. Общие свойства раствора – это свойства идеального раствора.
Идеальным называется раствор, образованный компонентами, имеющими строго одинаковые размеры частиц и строго одинаковую энергию межмолекулярного взаимодействия.
Основными законами, описывающими свойства растворов, являются: закон Генри, законы Рауля (1-й, 2-ой, 3-ий), осмотический закон Вант-Гоффа. Следует отметить, что эти законы выполняются только для разбавленных растворов.
Все растворы независимо от агрегатного состояния обладают способностью к диффузии. Диффузией называется свойство вещества равномерно распределяться по всему предоставленному ему объему. Скорость диффузии (скорость выравнивания концентрации по объему) в газах велика, в твердых телах при обычных температурах диффузия длится годы. В растворах диффузия протекает за десятки часов, для небольших порядка литра объемов раствора.
Если в сосуд налить концентрированный раствор, а сверху добавить чистый растворитель, то начнется процесс диффузии, как растворителя, так и растворенного вещества до полного выравнивания концентрации по всему объему.
Можно создать условия, когда диффузия идет только по растворителю. Для этого разделим раствор и чистый растворитель пленкой, через которую могут проходить только молекулы растворителя. Такие пленки называются полупроницаемыми мембранами.
Процесс односторонней диффузии растворителя через полупроницаемую мембрану называется осмосом.
При односторонней диффузии растворителя в раствор, объем последнего начинает увеличиваться, что влечет за собой увеличение гидростатического давления, которое препятствует диффузии растворителя. При некотором давлении наступает равновесие: сколько молекул растворителя проникло в раствор, столько же выталкивается из него увеличившимся давлением.
Равновесное давление раствора, препятствующее диффузии растворителя через полупроницаемую мембрану, называется осмотическим давлением.
Не нашли, что искали? Воспользуйтесь поиском: