ТОР 5 статей:
Методические подходы к анализу финансового состояния предприятия
Проблема периодизации русской литературы ХХ века. Краткая характеристика второй половины ХХ века
Характеристика шлифовальных кругов и ее маркировка
Служебные части речи. Предлог. Союз. Частицы
КАТЕГОРИИ:
- Археология
- Архитектура
- Астрономия
- Аудит
- Биология
- Ботаника
- Бухгалтерский учёт
- Войное дело
- Генетика
- География
- Геология
- Дизайн
- Искусство
- История
- Кино
- Кулинария
- Культура
- Литература
- Математика
- Медицина
- Металлургия
- Мифология
- Музыка
- Психология
- Религия
- Спорт
- Строительство
- Техника
- Транспорт
- Туризм
- Усадьба
- Физика
- Фотография
- Химия
- Экология
- Электричество
- Электроника
- Энергетика
Recombinant DNA Technology
http://darwin.nmsu.edu/~molbio/mcb520/lecture2.html
- Restriction enzymes
Isolated from various bacteria, restriction enzymes recognize short DNA sequences and cut the DNA molecules at those specific sites. (A natural biological function of these enzymes is toprotect bacteria by attacking viral and other foreign DNA.) Cuts yield either "staggered" or "sticky" ends (see figure) or "blunt" ends. Two pieces of DNA cut with the same enzyme, can be pasted together using another enzyme called "DNA ligase".

Some restriction enzymes cut the DNA very infrequently (rare-cutters), generating a small number of very large fragments (several thousand to a million base pairs). Most enzymes cut DNA more frequently, thus generating a large number of small fragments (less than a hundred to more than a thousand bp).
On average, restriction enzymes with
* 4- base recognition sites will yield pieces 256 bases long,
* 6- base recognition sites will yield pieces 4000 bases long, and
* 8- base recognition sites will yield pieces 64,000 bases long.
Why?
Since hundreds of different restriction enzymes have been characterized, DNA can be cut into many different small fragments.
- Agarose gel electrophoresis

DNA fragments that have been cut with REs can be separated based on their relative sizes by running them in a gel made of the polymer agarose, a jelly-like substance. DNA, which overall has a negative charge, is loaded into a well and current that runs from the negative to the positive poles is applied. DNA bands will separate with the smaller fragments running ahead of the bigger ones. A standard with DNA pieces of known sizes is run in parallel to estimate size of experimental DNAs. DNA is visualized by staining the gel with fluorescent ethidium bromide (EtBr). EtBr is hydrophobic substance that binds to the bases in double helix. Size can be more precisely established by interpolating the distance migrated by the experimental DNA into a plot charting size (in base-pairs) of the known standards in a semi-log scale on the Y-axis vs their migrated distance in the gel (in mm or cm or inches) in the X-axis. Resolution depends on the concentration of agarose used (usually 1%), the length of the gel and on the pattern of current applied.
- DNA sequencing
Two methods: the chemical method (Maxam and Gilbert) and the enzymatic or chain termination method (Sanger). The latter has become the standard procedure for sequencing DNA. This method is rapid, reliable, easy and accurate allowing the indirect determination of a proteinнs amino acid sequence by determining the nucleotide sequence of its coding DNA gene. Automation of this method has made possible the various genome sequencing projects, from small viruses, to E.coli to yeast and ultimately, it will allow the whole sequencing of the ~3 x 109 bps in the human genome. Explain method (Figure 7-8).
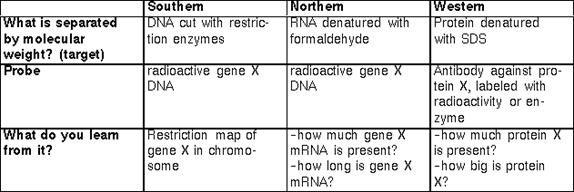
- Hybridization techniques

The principle of complementary base pairing introduced by Watson and Crick (A-T; G-C) is the basis of a series of techniques developed to detect, among a mixture of nuclei acids (DNA or RNA) immobilized onto a solid support, a specific target fragment or sequence. The target, in the appropiate conditions, will base-pair according to W/C rules with another piece of Nucleic Acid (the probe) that has its complementary nucleotide sequence. The probe is labelled for example with radioactive nucleotides, so that, after the proper washing out of unspecific products, the target can be detected by its binding to the labelled probe (using for example, autoradiography).
Depending on the nature of the target, hybridizations techniques include:
Southern bloting- DNA target/DNA probe-invented by the English Molecular Biologist E.M. Southern
Northern bloting- RNA target/DNA probe
Western blot-Protein target/antibody protein probe. The interaction between probe and target in a Western blot does not involve W/C base paring. Here, the biochemical complementary is between two protein surfaces and may involve non-covalent associations such as H-bonding or Ionic interactions.
SouthWestern blot-DNA target/Protein probe.
The following table summarize the major features of each hybridization technique.
In situ hybridization- either DNA or RNA probes are used to locate specific nuclei acid sequences in the cell, i.e. genes on chromosomes or mRNA if whatever compartment. Information about the temporal/spatial distribution of a mRNA can provide valuable insights about the function of the gene that encodes it. To increase spatial resolution non-radioactive methods of labelling the probes are preferred nowadays. However, some of these yield lower sensitivity than the radioactive probes.
- Genetic maps
 A genetic linkage map shows the RELATIVE locations of specific DNA markers along the chromosome. Any inherited physical or molecular characteristic that differs among individuals and is easily detectable in the laboratory is a potential genetic marker. Markers can be expressed DNA regions (genes) or DNA segments that have no known coding function but whose inheritance pattern can be followed. DNA sequence differences are especially useful markers because they are plentiful and easy to characterize precisely.
A genetic linkage map shows the RELATIVE locations of specific DNA markers along the chromosome. Any inherited physical or molecular characteristic that differs among individuals and is easily detectable in the laboratory is a potential genetic marker. Markers can be expressed DNA regions (genes) or DNA segments that have no known coding function but whose inheritance pattern can be followed. DNA sequence differences are especially useful markers because they are plentiful and easy to characterize precisely.
Markers must be polymorphic to be useful in mapping; that is, alternative forms must exist among individuals so that they are detectable among different members in family studies. Polymorphisms are variations in DNA sequence (mutations)that occur on average once every 300 to 500 bp. Variations within coding sequences can lead to observable changes, such as differences in eye color, blood type, and disease susceptibility. Most variations occur within non-coding regions and have little or no effect on an organisms appearance or function, yet they are detectable at the DNA level and can be used as markers. Examples of these types of markers include (1)restriction fragment length polymorphisms (RFLPs), which reflect sequence variations in DNA sites that can be cleaved by DNA restriction enzymes (see below), and (2)variable number of tandem repeat sequences, which are short repeated sequences that vary in the number of repeated units and, therefore, in length (a characteristic easily measured). The human genetic linkage map is constructed by observing how frequently two markers are inherited together which is determined by the recombination rate of the particular region. The closer the markers are to each other the more tightly-linked the less likely a recombination event will fall between and separate them. Recombination frequency thus provides an estimate of the distance between two markers.
On the genetic map, distances between markers are measured in terms of centimorgans (cM), named after the American geneticist Thomas Hunt Morgan. Two markers are said to be 1cM apart if they are separated by recombination 1% of the time. A genetic distance of 1cM is roughly equal to a physical distance of 1 million bp (1 Mb). Remember recombination frequency is not constant across the genome. It varies between regions of chromosomes. (Include Drosophila example, Genes V).
The value of the genetic map is that an inherited disease can be located on the map by following the inheritance of a DNA marker present in affected individuals (but absent in unaffected individuals), even though the molecular basis of the disease may not yet be understood nor the responsible gene identified. Genetic maps have been used to find the chromosomal location of several important disease genes, including cystic fibrosis, sickle cell disease, Tay-Sachs disease, fragile X syndrome, and myotonic dystrophy.
(How disease genes are found.. Insert Fig 6.10 Genes V).
Physical Maps
Physical maps vary in their degree of resolution. The lowest-resolution physical map is the chromosomal (cytogenetic) map, which is based on the distinctive banding patterns observed by light microscopy of stained chromosomes. A cDNA map shows the locations of expressed DNA regions (exons) on the chromosomal map. The more detailed cosmid contig map depicts the order of overlapping DNA fragments spanning the genome. A macrorestriction map describes the order and distance between enzyme cutting (cleavage) sites. The highest-resolution physical map is the complete elucidation of the DNA base-pair sequence of each chromosome in the human genome.
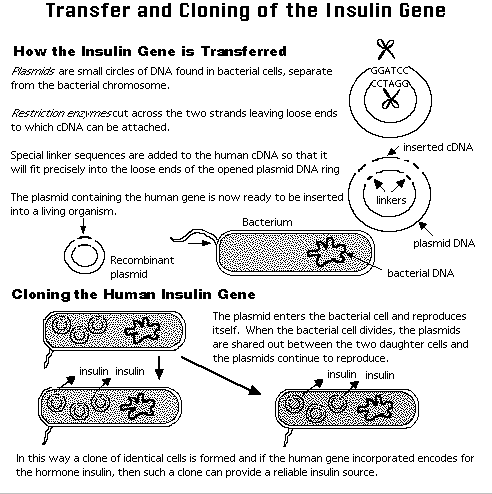
- Vectors, DNA cloning and libraries.
A clone describes a large number of identical cells or molecules with a single ancestral cell or molecule. Cloning vectors were developed from bacterial endogenous extrachromosomal or virus elements to shuttle or carry an inserted piece of DNA or a gene, for the purposes of producing more of it or of its protein product inside a bacterial host. RE are used to insert the foreign DNA into the vector. Types of vectors: plasmid, cosmids, yeast artificial chromosomes(YACs). Each vector was design to carry larger chunks of DNA with plasmids about 10 Kbp, Cosmids about 40Kbp and YAC about 1000Kbp, maximum. The example of the figure illustrates the cloning of one of the first human genes: the insulin gene.
- Cloning a Gene by Hybridization:
In this case, 'to clone the actin gene from humans' means "to end up with a plasmid which contains a fragment of human DNA which includes the actin gene". The usual starting point is a plasmid clone of the actin gene from another organism and human chromosomal DNA. DNA-DNA hybridization is usually used for this.
Although Southern blotting involves DNA-DNA hybridization, it is not a useful procedure for cloning a gene. If we were to cut human DNA with a restriction enzyme and run it on a Southern blot probed with a clone of the actin gene from an other organism, we could construct a restriction map of the human actin gene. However, we can not isolate the human actin gene DNA from either the gel or the filter because at each molecular weight on the gel, there are many bands of the same length but different sequences.
For this reason, separating the DNA fragments by molecular weight is unsuitable. Instead, we separate them by sequence - by making a LIBRARY, we end up with a collection of plasmids, physically separated, each containing a different fragment of human DNA. Here is a typical procedure: cloning the human gene for actin, given a clone of the yeast actin gene.
1) Isolate genomic (chromosomal) DNA from human cells.
2) Create a plasmid library of human DNA restriction fragments (genomic library). This results in a collection of bacterial colonies, each containing a different plasmid with a different inserted piece of human DNA.
3) Plate the colonies on agar plates and let them grow. These are called the master plates.
4) Press a piece of nitrocellulose onto each master plate and lift off. This leaves some of each colony on the plate and a replica on the filter.
5) Break open (lyse) the bacteria on the filter under conditions that make their plasmid DNA single-stranded, and bind the DNA onto the filter. There are now spots of single-stranded plasmid DNA on the filter. These spots correspond to the locations of the colonies of bacteria on the master plate.
6-9) "Screen" library by Southern hybridization, using a labeled restriction fragment of the yeast actin gene from the plasmid as a probe. In all likelihood, the stringency of the hybridization conditions would have to be "lowered" to allow stable annealing between the yeast and human actin sequences which, although similar, are surely not identical. The result will be a piece of X-ray film with a dark spot corresponding to a place on the filter where DNA from the human actin gene was present. This spot on the filter corresponds to a colony from the master plate.
10) Pick up some of the bacteria from the appropriate colony, grow them in broth and extract their plasmid DNA. It contains a fragment of human DNA containing the actin gene.
It is also possible to clone gene X using an antibody against the protein produced by gene X. In this case, you make an EXPRESSION LIBRARY - a library where the vector contains a strong E. coli promoter and an ATG codon. Cells containing these plasmids will produce large amounts of protein from whichever human gene they carry. If you prepare the filter replicas of the library as above, you can probe them with an antibody to find the colony that contains a plasmid that expresses protein X. This plasmid will contain gene X.
An expression library is a kind of cDNA Library. The starting material is the mRNA from a given tissue or cell. DNA copies of the mRNA(cDNA) are made using Reverse Transcriptase (RT), the same enzyme that allows retroviruses to replicate their RNA genome. The cDNA is the clone into a particular vector to generate the library. Subtractive hybridization is a technique to enrich for particular mRNA sequences prior to cDNA cloning. The goal is to eliminate sequences present in two samples and to be left with the sequences that are unique to a particular sample (i.e., cultured cells treated with a drug vs untreated cells. Most mRNAs, except those for the genes induced/repressed by the drug, will be present in these two populations. You would like to get rid of the former and enrich for the latter).
- DNA Amplification: PCR technology
Described as being to genes what Gutenberg's printing press was to the written word, PCR (or Polymerase chain reaction can amplify a desired DNA sequence of any origin (virus, bacteria, plant, or human) hundreds of millions of times in a matter of hours, a task that would have required several days with recombinant DNA technology. PCR is especially valuable because the reaction is highly specific, easily automated, and capable of amplifying minute amounts of sample. For these reasons, PCR has also had a major impact on clinical medicine, genetic disease diagnostics, forensic science, and evolutionary biology.
PCR is a process based on a heat-resistant DNA polymerase enzyme (purified from bacteria that lives in hot springs), which can synthesize a complementary strand to a given DNA strand in a mixture containing the 4 DNA bases and 2 DNA fragments (primers, each about 20 bases long) flanking the target sequence. The mixture is heated to separate the strands of double-stranded DNA containing the target sequence and then cooled to allow (1) the primers to find and bind to their complementary sequences on the separated strands and (2) the polymerase to extend the primers into new complementary strands. Repeated heating and cooling cycles multiply the target DNA exponentially, since each new double strand separates to become two templates for further synthesis. In about 1 hour, 20 PCR cycles can amplify the target by a million-fold.

| <== предыдущая лекция | | | следующая лекция ==> |
| | | Состав и пищеварительное действие желудочного сока |
Не нашли, что искали? Воспользуйтесь поиском: