ТОР 5 статей:
Методические подходы к анализу финансового состояния предприятия
Проблема периодизации русской литературы ХХ века. Краткая характеристика второй половины ХХ века
Характеристика шлифовальных кругов и ее маркировка
Служебные части речи. Предлог. Союз. Частицы
КАТЕГОРИИ:
- Археология
- Архитектура
- Астрономия
- Аудит
- Биология
- Ботаника
- Бухгалтерский учёт
- Войное дело
- Генетика
- География
- Геология
- Дизайн
- Искусство
- История
- Кино
- Кулинария
- Культура
- Литература
- Математика
- Медицина
- Металлургия
- Мифология
- Музыка
- Психология
- Религия
- Спорт
- Строительство
- Техника
- Транспорт
- Туризм
- Усадьба
- Физика
- Фотография
- Химия
- Экология
- Электричество
- Электроника
- Энергетика
Синтез жирных кислот из пальмитиновой кислоты
Удлинение жирных кислот. В ЭР происходит удлинение пальмитиновой кислоты с участием малонил-КоА. Последовательность реакций сходна с той, что происходит при синтезе пальмитиновой кислоты, однако в данном случае жирные кислоты связаны не с синтазой жирных кислот, а с КоА. Ферменты, участвующие в элонгации, могут использовать в качестве субстратов не только пальмитиновую, но и другие жирные кислоты, поэтому в организме могут синтезироваться не только стеариновая кислота, но и жирные кислоты с большим числом атомов углерода.Основной продукт элонгации в печени - стеариновая кислота (С 18:0), однако в ткани мозга образуется большое количество жирных кислот с более длинной цепью - от С20 до С24, которые необходимы для образования сфинголипидов и гликолипидов. В нервной ткани происходит синтез и других жирных кислот - α-гидроксикислот. Оксидазы со смешанными функциями гидроксилируют С22 и С24 кислоты с образованием лигноцериновой и цереброновой кислот, обнаруживаемых только в липидах мозга.
Образование двойных связей в радикалах жирных кислот. Включение двойных связей в радикалы жирных кислот называется десатурацией. Основные жирные кислоты, образующиеся в организме человека в результате десатурации - пальмитоо-леиновая (С16:1Δ9) и олеиновая (С18:1Δ9). Образование двойных связей в радикалах жирных кислот происходит в ЭР в реакциях с участием молекулярного кислорода, NADH и цитохрома b5. Ферменты десатуразы жирных кислот, имеющиеся в организме человека, не могут образовывать двойные связи в радикалах жирных кислот дистальнее девятого атома углерода, т.е. между девятым и метильным атомами углерода. Поэтому жирные кислоты семейства ω-3 и ω-6 не синтезируются в организме, являются незаменимыми и обязательно должны поступать с пищей, так как выполняют важные регуляторные функции. Для образования двойной связи в радикале жирной кислоты требуется молекулярный кислород, NADH, цитохром b5 и FAD-зависимая редуктаза цитохрома b5. Атомы водорода, отщепляемые от насыщенной кислоты, выделяются в виде воды. Один атом молекулярного кислорода включается в молекулу воды, а другой также восстанавливается до воды с участием электронов NADH, которые передаются через FADH2 и цитохром b
61.Химизм реакций β-окисления жирных кислот, энергетический итог.
β-Окисление - специфический путь катаболизма жирных кислот, при котором от карбоксильного конца жирной кислоты последовательно отделяется по 2 атома углерода в виде ацетил-КоА. Метаболический путь - β-окисление - назван так потому, что реакции окисления жирной кислоты происходят у β-углеродного атома. Реакции β-окисления и последующего окисления ацетил-КоА в ЦТК служат одним из основных источников энергии для синтеза АТФ по механизму окислительного фосфорилирования. β-Окисление жирных кислот происходит только в аэробных условиях.
Активация жирных кислот. Перед тем, как вступить в различные реакции, жирные кислоты должны быть активированы, т.е. связаны макроэргической связью с коферментом А:
RCOOH + HSKoA + АТФ → RCO ~ КоА + АМФ + PPi.
Реакцию катализирует фермент ацил-КоА син-тетаза. Выделившийся в ходе реакции пирофосфат гидролизуется ферментом пирофосфатазой: Н4Р2О7 + Н2О → 2 Н3РО4. Выделение энергии при гидролизе макроэргической связи пирофосфата смещает равновесие реакции вправо и обеспечивает полноту протекания реакции активации. Ацил-КоА синтетазы находятся как в цитозоле, так и в матриксе митохондрий. Эти ферменты отличаются по специфичности к жирным кислотам с различной длиной углеводородной цепи. Жирные кислоты с короткой и средней длиной цепи (от 4 до 12 атомов углерода) могут проникать в матрикс митохондрий путём диффузии. Активация этих жирных кислот происходит в матриксе митохондрий. Жирные кислоты с длинной цепью, которые преобладают в организме человека (от 12 до 20 атомов углерода), активируются ацил-КоА синтетазами, расположенными на внешней мембране митохондрий.
Транспорт жирных кислот с длинной углеводородной цепью в митохондриях. β-Окисление жирных кислот, происходит в матриксе митохондрий, поэтому после активации жирные кислоты должны транспортироваться внутрь митохондрий. Жирные кислоты с длинной углеводородной цепью переносятся через плотную внутреннюю мембрану митохондрий с помощью карнитина. Карнитин поступает с пищей или синтезируется из незаменимых аминокислот лизина и метионина. В реакциях синтеза карнитина участвует витамин С (аскорбиновая кислота).В наружной мембране митохондрий находится фермент карнитинацилтрансфераза I (карнитин-пальмитоилтрансфераза I), катализирующий реакцию с образованием ацилкарнитина. Образовавшийся ацилкарнитин проходит через межмембранное пространство к наружной стороне внутренней мембраны и транспортируется с помощью карнитинацилкарнитинтранс-локазы на внутреннюю поверхность внутренней мембраны митохондрий, где фермент карнитинацилтрансфераза II катализирует перенос ацила на внутримитохондриальный КоА. Таким образом, ацил-КоА становится доступным для ферментов β-окисления. Свободный карнитин возвращается на цитозольную сторону внутренней мембраны митохондрий той же транслоказой. На внутренней поверхности внутренней мембраны находится фермент карнитинацил трансфераза II, катализирующий обратный перенос ацила с карнитина на внутримитохондриальный КоА. После этого ацил-КоА включается в реакции β-окисления. 
β- Окисление жирных кислот - специфический путь катаболизма жирных кислот, протекающий в матриксе митохондрий только в аэробных условиях и заканчивающийся образованием ацетил-КоА. Водород из реакций β-окисления поступает в ЦПЭ, а ацетил-КоА окисляется в цитратном цикле, также поставляющем водород для ЦПЭ. Поэтому β-окисление жирных кислот - важнейший метаболический путь, обеспечивающий синтез АТФ в дыхательной цепи.

β-Окисление начинается с дегидрирования ацил-КоА FAD-зависимой ацил-КоА дегидрогеназой с образованием двойной связи между α- и β-атомами углерода в продукте реакции - еноил-КоА. Восстановленный в этой реакции кофермент FADH2 передаёт атомы водорода в ЦПЭ на кофермент Q. В результате синтезируются 2 молекулы АТФ. В следующей реакции р-окисления по месту двойной связи присоединяется молекула воды таким образом, что ОН-группа находится у β-углеродного атома ацила, образуя β-гидроксиацил-КоА. Затем β-гидроксиацил-КоА окисляется NАD+-зависимой дегидрогеназой. Восстановленный NADH, окисляясь в ЦПЭ, обеспечивает энергией синтез 3 молекул АТФ. Образовавшийся β-кетоацил-КоА подвергается тиолитическому расщеплению ферментом тиолазой, так как по месту разрыва связи С-С через атом серы присоединяется молекула кофермента А. В результате этой последовательности из 4 реакций от ацил-КоА отделяется двухуглеродный остаток - ацетил-КоА. Жирная кислота, укороченная на 2 атома углерода, опять проходит реакции дегидрирования, гидратации, дегидрирования, отщепления ацетил-КоА. Эту последовательность реакций обычно называют "циклом β-окисления", имея в виду, что одни и те же реакции повторяются с радикалом жирной кислоты до тех пор, пока вся кислота не превратится в ацетильные остатки.
Продуктами каждого цикла β-окисления являются FADH2, NADH и ацетил-КоА. Хотя реакции в каждом "цикле" одни и те же, остаток кислоты, который входит в каждый последующий цикл, короче на 2 углеродных атома. В последнем цикле окисляется жирная кислота из 4 атомов углерода, поэтому образуются 2 молекулы ацетил-КоА, а не 1, как в предыдущих. Суммарное уравнение β-окисления, например пальмитоил-КоА может быть представлено таким образом:
С15Н31СО-КоА + 7 FAD + 7 NAD+ + 7 HSKoA → 8 СН3-СО-КоА + 7 FADH2 + 7 (NADH + H+).
Если рассчитывать выход АТФ при окислении пальмитиновой кислоты то из общей суммы молекул АТФ необходимо вычесть 2 молекулы, так как на активацию жирной кислоты тратится энергия 2 макроэргических связей. Следовательно, энергетический выход равен:
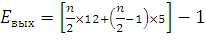
где n/2 – количество молекул Ацетил-КоА
n – количество С-атомов
12 – количество АТФ, получаемое при окислении Ацетил-КоА
n/2-1 – количество циклов β-окисления
5 – количество молекул АТФ, образовавшихся в цикле за счет 2 реакций дегидрирования
-1 – затрата одной АТФ
62. Биосинтез и использование кетоновых тел в качестве источников энергии
При голодании, длительной физической работе и в случаях, когда клетки не получают достаточного количества глюкозы, жирные кислотыиспользуются многими тканями как основной источник энергии. В отличие от других тканей мозг и другие отделы нервной ткани практически не используют жирные кислоты в качестве источника энергии. В печени часть жирных кислот превращается в кетоновые тела, которые окисляются мозгом, нервной тканью, мышцами, обеспечивая достаточное количество энергии для синтеза АТФ и уменьшая потребление глюкозы. К кетоновымтелам относят β-гидроксибутират, ацетоацетат и ацетон. Первые две молекулы могут окисляться в тканях, обеспечивая синтез АТФ. Ацетон образуется только при высоких концентрациях кетоновых тел в крови и, выделяясь с мочой, выдыхаемым воздухом и потом, позволяет организму избавляться от избытка кетоновых тел.
Синтез кетоновых тел в печени. При низком соотношении инсулин/глюкагон в крови в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, поэтому увеличивается скорость β-окисления. Скорость реакций ЦТК в этих условиях снижена, так как оксалоацетат используется для глюконеогенеза. В результате скорость образования ацетил-КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза кетоновых тел. Синтез кетоновых тел происходит только в митохондриях печени.

Синтез кетоновых тел начинается с взаимодействия двух молекул ацетил-КоА, которые под действием фермента тиолазы образуют ацетоацетил-КоА. С ацетоацетил-КоА взаимодействует третья молекула ацетил-КоА, образуя 3-гидрокси-3-метилглутарил-КоА (ГМГ-КоА). Эту реакцию катализирует фермент ГМГ-КоА-синтаза. Далее ГМГ-КоА-лиаза катализирует расщепление ГМГ-КоА на свободный ацетоацетат и ацетил-КоА. Ацетоацетат может выделяться в кровь или превращаться в печени в другое кетоновое тело - β-гидроксибутират путём восстановления. В клетках печени при активном β-окислении создаётся высокая концентрация NADH. Это способствует превращению большей части ацетоацетата в β-гидроксибутират, поэтому основное кетоновое тело в крови - именно β-гидроксибутират. При голодании для многих тканей жирные кислоты и кетоновые тела становятся основными топливными молекулами. Глюкоза используется в первую очередь нервной тканью и эритроцитами. При высокой концентрации ацетоацетата часть его неферментативно декарбоксилируется, превращаясь в ацетон. Ацетон не утилизируется тканями, но выделяется с выдыхаемым воздухом и мочой. Таким путём организм удаляет избыточное количество кетоновых тел, которые не успевают окисляться, но, являясь водорастворимыми кислотами, вызывают ацидоз. При длительном голодании кетоновые тела становятся основным источником энергии для скелетных мышц, сердца и почек. Таким образом глюкоза сохраняется для окисления в мозге и эритроцитах. Уже через 2-3 дня после начала голодания концентрация кетоновых тел в крови достаточна для того, чтобы они проходили в клетки мозга и окислялись, снижая его потребности в глюкозе. β-Гидроксибутират, попадая в клетки, дегидрируется NAD-зависимой дегидрогеназой и превращается в ацетоацетат. Ацетоацетат активируется, взаимодействуя с сук-цинил-КоА - донором КоА:
Ацетоацетат + Сукцинил-КоА → Ацетоацетил- КоА + Сукцинат.
Реакцию катализирует сукцинил-КоА-ацето-ацетат-КоА-трансфераза. Этот фермент не синтезируется в печени, поэтому печень не использует кетоновые тела как источники энергии, а производит их "на экспорт". Кетоновые тела - хорошие топливные молекулы; окисление одной молекулы β-гидроксибутирата до СО2 и Н2О обеспечивает синтез 27 молекул АТФ. Эквивалент одной макроэргической связи АТФ (в молекуле сукцинил-КоА) используется на активацию ацетоацетата, поэтому суммарный выход АТФ при окислении одной молекулы β-гидроксибутирата - 26 молекул.
6З.Пищевые жиры и их переваривание. Всасывание продуктов переваривания. Нарушение переваривания и всасывания. Ресинтез триацилглицеринов в стенке кишечника.
Жиры составляют до 90% липидов, поступающих с пищей. Переваривание жиров происходит в тонком кишечнике, однако уже в желудке небольшая часть жиров гидролизуется под действием " липазы языка ". Этот фермент синтезируется железами на дорсальной поверхности языка и относительно устойчив при кислых значениях рН желудочного сока. Поэтому он действует в течение 1-2 ч на жиры пищи в желудке. Однако вклад этой липазы в переваривание жиров у взрослых людей незначителен. Основной процесс переваривания происходит в тонкой кишке.
Так как жиры - нерастворимые в воде соединения, то они могут подвергаться действию ферментов, растворённых в воде только на границе раздела фаз вода/жир. Поэтому действию панкреатической липазы, гидролизующей жиры, предшествует эмульгирование жиров. Эмульгирование (смешивание жира с водой) происходит в тонком кишечнике под действием солей жёлчных кислот. Жёлчные кислоты синтезируются в печени из холестерола и секретируются в жёлчный пузырь. Содержимое жёлчного пузыря - жёлчь. Это вязкая жёлто-зелёная жидкость, содержащая главным образом жёлчные кислоты; в небольшом количестве имеются фосфолипиды и холестерол. Жёлчные кислоты представляют собой в основном конъюгированные жёлчные кислоты: таурохолевую, гликохолевую и другие. После приёма жирной пищи жёлчный пузырь сокращается и жёлчь изливается в просвет двенадцатиперстной кишки. Жёлчные кислоты действуют как детергенты, располагаясь на поверхности капель жира и снижая поверхностное натяжение. В результате крупные капли жира распадаются на множество мелких, т.е. происходит эмульгирование жира. Эмульгирование приводит к увеличению площади поверхности раздела фаз жир/вода, что ускоряет гидролиз жира панкреатической липазой. Эмульгированию способствует и перистальтика кишечника. Переваривание жиров - гидролиз жиров панкреатической липазой. Оптимальное значение рН для панкреатической липазы ≈8 достигается путём нейтрализации кислого содержимого, поступающего из желудка, бикарбонатом, выделяющимся в составе сока поджелудочной железы:
Н+ + НСО3- → Н2СО3 → Н2О + СО2 ↑.
Выделяющийся углекислый газ способствует дополнительному перемешиванию содержимого тонкой кишки.
Панкреатическая липаза выделяется в полость тонкой кишки из поджелудочной железы вместе с белком колипазой. Колипаза попадает в полость кишечника в неактивном виде и частичным протеолизом под действием трипсина превращается в активную форму. Колипаза своим гидрофобным доменом связывается с поверхностью мицеллы эмульгированного жира. Другая часть молекулы способствует формированию такой конформации панкреатической липазы, при которой активный центр фермента максимально приближен к своим субстратам - молекулам жиров поэтому скорость реакции гидролиза жира резко возрастает
Панкреатическая липаза гидролизует жиры преимущественно в положениях 1 и 3 поэтому основными продуктами гидролиза являются свободные жирные кислоты и 2-моноацилглицеролы (β-моноацилглицеролы). Молекулы 2-моноацилглицеролов также обладают детергентными свойствами и способствуют эмульгированию жира.
Образование смешанных мицелл и всасывание продуктов гидролиза. Продукты гидролиза липидов - жирные кислоты с длинным углеводородным радикалом, 2-моноацилглицеролы, холестерол, а также соли жёлчных кислот образуют в просвете кишечника структуры, называемые смешанными мицеллами. Смешанные мицеллы построены таким образом, что гидрофобные части молекул обращены внутрь мицеллы, а гидрофильные - наружу, поэтому мицеллы хорошо растворяются в водной фазе содержимого тонкой кишки. Стабильность мицелл обеспечивается в основном солями жёлчных кислот. Мицеллы сближаются со щёточной каймой клеток слизистой оболочки тонкого кишечника, и липидные компоненты мицелл диффундируют через мембраны внутрь клеток. Вместе с продуктами гидролиза липидов всасываются жирорастворимые витамины A, D, Е, К и соли жёлчных кислот. Наиболее активно соли жёлчных кислот всасываются в подвздошной кишке. Жёлчные кислоты далее попадают через воротную вену в печень, из печени вновь секретируются в жёлчный пузырь и далее опять участвуют в эмульгировании жиров. Этот путь жёлчных кислот называют " энтерогепатическая циркуляция". Каждая молекула жёлчных кислот за сутки проходит 5- 8 циклов, и около 5% жёлчных кислот выделяется с фекалиями. Всасывание жирных кислот со средней длиной цепи, образующихся, например, при переваривании липидов молока, происходит без участия смешанных мицелл. Эти жирные кислоты из клеток слизистой оболочки тонкого кишечника попадают в кровь, связываются с белком альбумином и транспортируются в печень.
Ресинтез жиров в слизистой оболочке тонкого кишечника. После всасывания продуктов гидролиза жиров жирные кислоты и 2-моноацилглицеролы в клетках слизистой оболочки тонкого кишечника включаются в процесс ресинтеза с образованием триацилглицеролов Жирные кислоты вступают в реакцию этерификации только в активной форме в виде производных коэнзима А, поэтому первая стадия ресинтеза жиров - реакция активации жирной кислоты:
HS КоА + RCOOH + АТФ → R-CO ~ КоА + АМФ + Н4Р2О7.
Реакция катализируется ферментом ацил-КоА-синтетазой (тиокиназой). Затем ацил~КоА участвует в реакции этерификации 2-моноацилглицерола с образованием сначала диацилгли-церола, а затем триацилглицерола. Реакции ресинтеза жиров катализируют ацилтранеферазы. В реакциях ресинтеза жиров участвуют, как правило, только жирные кислоты с длинной углеводородной цепью. В ресинтезе жиров участвуют не только жирные кислоты, всосавшиеся из кишечника, но и жирные кислоты, синтезированные в организме, поэтому по составу ре-синтезированные жиры отличаются от жиров, полученных с пищей. Однако возможности "адаптировать" в процессе ресинтеза состав пищевых жиров к составу жиров организма человека ограничены, поэтому при поступлении с пищей жиров с необычными жирными кислотами, например бараньего жира, в адипоцитах появляются жиры, содержащие кислоты, характерные для бараньего жира (насыщенные разветвлённые жирные кислоты). В клетках слизистой оболочки кишечника происходит активный синтез глицерофосфолипидов, необходимых для формирования структуры липопротеинов - транспортных форм липидов в крови.
Нарушения переваривания и всасывания жиров. Нарушение переваривания жиров может быть следствием нескольких причин. Одна из них - нарушение секреции жёлчи из жёлчного пузыря при механическом препятствии оттоку жёлчи. Это состояние может быть результатом сужения просвета жёлчного протока камнями, образующимися в жёлчном пузыре, или сдавлением жёлчного протока опухолью, развивающейся в окружающих тканях. Уменьшение секреции жёлчи приводит к нарушению эмульгирования пищевых жиров и, следовательно, к снижению способности панкреатической липазы гидролизовать жиры.
Нарушение секреции сока поджелудочной железы и, следовательно, недостаточная секреция панкреатической липазы также приводят к снижению скорости гидролиза жиров. В обоих случаях нарушение переваривания и всасывания жиров приводит к увеличению количества жиров в фекалиях - возникает стеаторе я (жирный стул). В норме содержание жиров в фекалиях составляет не более 5%. При стеаторее нарушается всасывание жирорастворимых витаминов (A, D, E, К) и незаменимых жирных кислот, поэтому при длительно текущей стеаторее развивается недостаточность этих незаменимых факторов питания с соответствующими клиническими симптомами. При нарушении переваривания жиров плохо перевариваются и вещества нелипидной природы, так как жир обволакивает частицы пищи и препятствует действию на них ферментов.
64.Образование хиломикронов и транспорт жиров. Роль апопротеинов в составе хиломикронов. Липопротеинлипаза.
Липиды в водной среде (а значит, и в крови) нерастворимы, поэтому для транспорта липидов кровью в организме образуются комплексы липидов с белками - липопротеины. Все типы липопротеинов имеют сходное строение - гидрофобное ядро и гидрофильный слой на поверхности. Гидрофильный слой образован белками, которые называют апопротеинами, и амфифильными молекулами липидов - фосфолипидами и холестеролом. Гидрофильные группы этих молекул обращены к водной фазе, а гидрофобные части - к гидрофобному ядру липопротеина, в котором находятся транспортируемые липиды. Некоторые апопротеины интегральные и не могут быть отделены от липопротеина, а другие могут свободно переноситься от одного типа липопротеина к другому. Апопротеины выполняют несколько функций:
· формируют структуру липопротеинов;
· взаимодействуют с рецепторами на поверхности клеток и таким образом определяют, какими тканями будет захватываться данный тип липопротеинов;
· служат ферментами или активаторами ферментов, действующих на липопротеины.
В организме синтезируются следующие типы липопротеинов: хиломикроны (ХМ), липопротеины очень низкой плотности (ЛПОНП), липопротеины промежуточной плотности (ЛППП), липопротеины низкой плотности (ЛПНП) и липопротеины высокой плотности (ЛПВП). Каждый из типов ЛП образуется в разных тканях и транспортирует определённые липиды. Например, ХМ транспортируют экзогенные (пищевые жиры) из кишечника в ткани, поэтому триацилглицеролы составляют до 85% массы этих частиц. ЛП хорошо растворимы в крови, не коалесцируют, так как имеют небольшой размер и отрицательный заряд на поверхности. Некоторые ЛП легко проходят через стенки капилляров кровеносных сосудов и доставляют липиды к клеткам. Большой размер ХМ не позволяет им проникать через стенки капилляров, поэтому из клеток кишечника они сначала попадают в лимфатическую систему и потом через главный грудной проток вливаются в кровь вместе с лимфой. Жиры, образовавшиеся в результате ресинтеза в клетках слизистой оболочки кишечника, упаковываются в ХМ. Основной апопротеин в составе ХМ - белок апоВ-48. В лимфе и крови с ЛПВП на ХМ переносятся апопротеины Е (апоЕ) и С-П (апоС-П); ХМ превращаются в "зрелые". ХМ имеют довольно большой размер, поэтому после приёма жирной пищи они придают плазме крови опалесцирующий, похожий на молоко, вид. ХМ транспортируют жир к различным тканям, где он утилизируется, поэтому концентрация ХМ в крови постепенно снижается, и плазма опять становится прозрачной. ХМ исчезают из крови в течение нескольких часов.
| Типы липопротеинов | Хиломикроны (ХМ) | ЛПОНП | ЛППП | ЛПНП | ЛПВП |
| Состав, % | |||||
| Белки | |||||
| ФЛ | |||||
| ХС | |||||
| ЭХС | |||||
| ТАГ | |||||
| Функции | Транспорт липидов из клеток кишечника(экзогенных липидов) | Транспорт липидов, синтезируемых в печени (эндогенных липидов) | Промежуточная форма превращения ЛПОНП в ЛПНП под действием фермента ЛП-липазы | Транспорт холестерола в ткани | Удаление избытка холестерола из клеток и других липопротеинов. Донор апопротеинов А, С-П |
| Место образования | Эпителий тонкого кишечника | Клетки печени | Кровь | Кровь (из ЛПОНП и ЛППП) | Клетки печени - ЛПВП-пред-шественники |
| Плотность, г/мл | 0,92-0,98 | 0,96-1,00 | 1,00-1,06 | 1,06-1,21 | |
| Диаметр частиц, нМ | Больше 120 | 30-100 | 21-100 | 7-15 | |
| Основные аполипопротеины | В-48 С-П Е | В-100 С-П Е | В-100 Е | В-100 | A-I С-II Е |
Действие липопротеинлипазы на ХМ. В крови триацилглицеролы, входящие в состав зрелых ХМ, гидролизуются ферментом липопротеин-липазой, или ЛП-липазой. ЛП-липаза связана с гепарансульфатом (гетерополисахаридом), находящимся на поверхности эндотелиальных клеток, выстилающих стенки капилляров кровеносных сосудов. ЛП-липаза гидролизует молекулы жиров до глицерола и 3 молекул жирных кислот. На поверхности ХМ различают 2 фактора, необходимых для активности ЛП-липазы - апоС-П и фосфолипиды. АпоС-П активирует этот фермент, а фосфолипиды участвуют в, связывании фермента с поверхностью ХМ. ЛП-липаза синтезируется в клетках многих тканей: жировой, мышечной, в лёгких, селезёнке, клетках лактирующей молочной железы. Изоферменты ЛП-липазы в разных тканях отличаются по значению Кm: ЛП-липаза жировой ткани имеет в 10 раз более высокое значение Кm, чем, например, ЛП-липаза сердца, поэтому гидролиз жиров ХМ в жировой ткани происходит в абсорбтивный период. Жирные кислоты поступают в адипоциты и используются для синтеза жиров. В постабсорбтивном состоянии, когда количество жиров в крови снижается, ЛП-липаза сердечной мышцы продолжает гидролизовать жиры в составе ЛПОНП, которые присутствуют в крови в небольшом количестве, и жирные кислоты используются этой тканью как источники энергии, даже при низкой концентрации жиров в крови. ЛП-липазы нет в печени, но на поверхности клеток этого органа имеется другой фермент - печёночная липаза, не действующая на зрелые ХМ, но гидролизующая жиры в ЛППП, которые образуются из ЛПОНП.В результате действия ЛП-липазы на жиры ХМ образуются жирные кислоты и глицерол. Основная масса жирных кислот проникает в ткани. В жировой ткани в абсорбтивный период жирные кислоты депонируются в виде триацилглицеролов, в сердечной мышце и работающих скелетных мышцах используются как источник энергии. Другой продукт гидролиза жиров, глицерол, растворим в крови, транспортируется в печень, где в абсорбтивный период может быть использован для синтеза жиров В результате действия ЛП-липазы на ХМ количество жиров в них снижается на 90%, уменьшаются размеры частиц, апопротеин С-П переносится обратно на ЛПВП. Образовавшиеся частицы называются остаточными ХМ. Они содержат в себе фосфолипиды, холестерол, жирорастворимые витамины и апопротеины В-48 и Е. Остаточные ХМ захватываются гепатоцитами, которые имеют рецепторы, взаимодействующие с этими апопротеинами. Путём эндоцитоза остаточные ХМ попадают внутрь клеток, и ферментами лизосом белки и липиды гидролизуются, а затем утилизируются. Жирорастворимые витамины и экзогенный холестерол используются в печени или транспортируются в другие ткани.
65.Биосинтез жиров в печени из углеводов. Структура и состав транспортных липопротеинов крови.
В жировой ткани для синтеза жиров используются в основном жирные кислоты, освободившиеся при гидролизе жиров ХМ и ЛПОНП. Жирные кислоты поступают в адипоциты, превращаются в производные КоА и взаимодействуют с глицерол-3-фосфатом, образуя сначала лизофосфатидную кислоту, а затем фосфатидную. Фосфатидная кислота после дефосфорилирования превращается в диацилглицерол, который ацилируется с образованием триацилглицерола.
Кроме жирных кислот, поступающих в адипоциты из крови, в этих клетках идёт и синтез жирных кислот из продуктов распада глюкозы. В адипоцитах для обеспечения реакций синтеза жира распад глюкозы идёт по двум путям: гликолиз, обеспечивающий образование глицерол-3-фосфата и ацетил-КоА, и пентозофосфатный путь, окислительные реакции которого обеспечивают образование NADPH, служащего донором водорода в реакциях синтеза жирных кислот.
Молекулы жиров в адипоцитах объединяются в крупные жировые капли, не содержащие воды, и поэтому являются наиболее компактной формой хранения топливных молекул. Подсчитано, что, если бы энергия, запасаемая в жирах, хранилась в форме сильно гидратированных молекул гликогена, то масса тела человека увеличилась бы на 14-15 кг. Печень - основной орган, где идёт синтез жирных кислот из продуктов гликолиза. В гладком ЭР гепатоцитов жирные кислоты активируются и сразу же используются для синтеза жиров, взаимодействуя с глицерол-3-фосфатом. Как и в жировой ткани, синтез жиров идёт через образование фосфатидной кислоты. Синтезированные в печени жиры упаковываются в ЛПОНП и секретируются в кровь

| Типы липопротеинов | Хиломикроны (ХМ) | ЛПОНП | ЛППП | ЛПНП | ЛПВП |
| Состав, % | |||||
| Белки | |||||
| ФЛ | |||||
| ХС | |||||
| ЭХС | |||||
| ТАГ | |||||
| Функции | Транспорт липидов из клеток кишечника(экзогенных липидов) | Транспорт липидов, синтезируемых в печени (эндогенных липидов) | Промежуточная форма превращения ЛПОНП в ЛПНП под действием фермента ЛП-липазы | Транспорт холестерола в ткани | Удаление избытка холестерола из клеток и других липопротеинов. Донор апопротеинов А, С-П |
| Место образования | Эпителий тонкого кишечника | Клетки печени | Кровь | Кровь (из ЛПОНП и ЛППП) | Клетки печени - ЛПВП-пред-шественники |
| Плотность, г/мл | 0,92-0,98 | 0,96-1,00 | 1,00-1,06 | 1,06-1,21 | |
| Диаметр частиц, нМ | Больше 120 | 30-100 | 21-100 | 7-15 | |
| Основные аполипопротеины | В-48 С-П Е | В-100 С-П Е | В-100 Е | В-100 | A-I С-II Е |
В состав ЛПОНП, кроме жиров, входят холестерол, фосфолипиды и белок - апоВ-100. Это очень "длинный" белок, содержащий 11 536 аминокислот. Одна молекула апоВ-100 покрывает поверхность всего липопротеина.
ЛПОНП из печени секретируются в кровь, где на них, как и на ХМ, действует ЛП-липаза. Жирные кислоты поступают в ткани, в частности в адипоциты, и используются для синтеза жиров. В процессе удаления жиров из ЛПОНП под действием ЛП-липазы ЛПОНП сначала превращаются в ЛППП, а затем в ЛПНП. В ЛПНП основными липидными компонентами служат холестерол и его эфиры, поэтому ЛПНП являются липопротеинами, доставляющими холестерол в периферические ткани. Глицерол, освободившийся из липопротеинов, кровью транспортируется в печень, где опять может использоваться для синтеза жиров.

66. Депонирование и мобилизация жиров в жировой ткани. Регуляция синтеза и мобилизации жиров. Роль инсулина, глюкагона и адреналина.
Какой процесс будет преобладать в организме - синтез жиров (липогенез) или их распад (липолиз), зависит от поступления пищи и физической активности. В абсорбтивном состоянии под действием инсулина происходит липогенез, в постабсорбтивном состоянии - липолиз, активируемый глюкагоном. Адреналин, секреция которого увеличивается при физической активности, также стимулирует липолиз.
Регуляция синтеза жиров. В абсорбтивный период при увеличении соотношения инсулинглюкагон в печени активируется синтез жиров. В жировой ткани индуцируется синтез ЛП-липазы в адипоцитах и осуществляется её экспонирование на поверхность эндотелия; следовательно, в этот период увеличивается поступление жирных кислот в адипоциты. Одновременно инсулин активирует белки-переносчики глюкозы - ГЛЮТ-4. Поступление глюкозы в адипоциты и гликолиз также активируются. В результате образуются все необходимые компоненты для синтеза жиров: глицерол-3-фосфат и активные формы жирных кислот. В печени инсулин, действуя через различные механизмы, активирует ферменты путём дефосфорилирования и индуцирует их синтез. В результате увеличиваются активность и синтез ферментов, участвующих в превращении части глюкозы, поступающей с пищей, в жиры. Это - регуляторные ферменты гликолиза, пируватдегидрогеназный комплекс и ферменты, участвующие в синтезе жирных кислот из ацетил-КоА. Результат действия инсулина на обмен углеводов и жиров в печени - увеличение синтеза жиров и секреция их в кровь в составе ЛПОНП. ЛПОНП доставляют жиры в капилляры жировой ткани, где действие ЛП-липазы обеспечивает быстрое поступление жирных кислот в адипоциты, где они депонируются в составе триацилглицеринов. Запасание жиров в жировой ткани - основная форма депонирования источников энергии в организме человека. Запасы жиров в организме человека массой 70 кг составляют 10 кг, но у многих людей количество жиров может быть значительно больше. Жиры образуют в адипоцитах жировые вакуоли. Жировые вакуоли иногда заполняют значительную часть цитоплазмы. Скорость синтеза и мобилизации подкожного жира происходит неравномерно в разных частях организма, что связано с неодинаковым распределением рецепторов гормонов на адипоцитах.
Регуляция мобилизации жиров. Мобилизация депонированных жиров стимулируется глюкагоном и адреналином и, в меньшей степени, некоторыми другими гормонами (соматотроп-ным, кортизолом). В постабсорбтивный период и при голодании глюкагон, действуя на адипоциты через аденилатциклазную систему, активирует протеинкиназу А, которая фосфо-рилирует и, таким образом, активирует гормончувствительную липазу, что инициирует липо-лиз и выделение жирных кислот и глицерина в кровь. При физической активности увеличивается секреция адреналина, который действует через β-адренергические рецепторы адипоцитов, активирующие аденилатциклазную систему. В настоящее время обнаружено 3 типа β-рецепторов: β1, β2, β3, активация которых приводит к липолитическому действию. К наибольшему липолитическому действию приводит активация β3-рецепторов. Адреналин одновременно действует и на α2-рецепторы адипоцитов, связанные с ингибирующим G-белком, что инактивирует аденилатциклазную систему. Вероятно, действие адреналина двояко: при низких концентрациях в крови преобладает его антилиполитическое действие через α2-рецепторы, а при высокой - преобладает липолитическое действие через β-рецепторы.Для мышц, сердца, почек, печени при голодании или физической работе жирные кислоты становятся важным источником энергии. Печень перерабатывает часть жирных кислот в кетоновые тела, используемые мозгом, нервной тканью и некоторыми другими тканями как источники энергии. В результате мобилизации жиров концентрация жирных кислот в крови увеличивается приблизительно в 2 раза, однако абсолютная концентрация жирных кислот в крови невелика даже в этот период. Т1/2 жирных кислот в крови тоже очень мал (менее 5 мин), что означает существование быстрого потока жирных кислот из жировой ткани к другим органам. Когда постабсорбтивный период сменяется аборбтивным, инсулин активирует специфическую фосфатазу, которая дефосфорилирует гормончувствительную липазу, и распад жиров останавливается.
| синтез гликогена |
| глюкокортикоиды |
| гликогенез |
| мобилизация гликогена в печени |
| глюкагон, адреналин |
| протеиногенез |
| всасывание |
| другие моносахариды |
| инсулин |
| липогенез |
| инсулин |
| глюкагон |
| Кровоток |
67.Основные фосфолипиды и гликолипиды тканей человека (глицерофосфолипиды, сфингофосфолипиды, гликоглицеролипиды, гликосфиголипиды). Представление о биосинтезе и катаболизме этих соединений.
Синтез фосфатидилхолинов, фосфатидилэтаноламинов и фосфатидилсеринов. Начальные этапы синтеза глицерофосфолипидов и жиров происходят одинаково до образования фосфатидной кислоты. Фосфатидная кислота может синтезироваться двумя разными путями: через глицеральдегид-3-фосфат и через дигидроксиацетонфосфат

На следующем этапе фосфатидаза отщепляет от фосфатидной кислоты фосфатный остаток, в результате чего образуется диацилглицерол. Дальнейшие превращения диацилглицерола также могут идти разными путями. Один из вариантов - образование активной формы "полярной головки" фосфолипида: холин, серии или этаноламин превращаются в ЦДФ-холин, ЦДФ-серин или ЦДФ-этаноламин. Далее диацилглицерол взаимодействует с ЦМФ-производными, при этом выделяется ЦМФ, и образуется соответствующий фосфолигид, например фосфатидилхолин. Между глицерофосфолипидами возможны различные взаимопревращения. Фосфатидилхолин может образовываться и другим путём: из фосфатидилэтаноламина, получая последовательно 3 метальные группы от SAM. Фосфатидилсерин может превращаться в фосфатидилэтаноламин путём декарбоксилирования. Фосфатидилэтаноламин может превращаться в фосфатидилсерин путём обмена этаноламина на серии.
Сфинголипиды - производные церамида, образующегося в результате соединения аминоспирта сфингозина и жирной кислоты. В группу сфинголипидов входят сфингомиелины и гликосфинголипиды фингомиелины находятся в мембранах клеток различных тканей, но наибольшее их количество содержится в нервной ткани. Сфингомиелины миелиновых оболочек содержат в основном жирные кислоты с длинной цепью: лигноцери-новую (24:0) и нервоновую (24:1) кислоты, а сфингомиелин серого вещества мозга содержит преимущественно стеариновую кислоту.

Гликосфинголипиды - гликолипиды, в состав которых входят церамид и один или несколько остатков углеводов, и сиаловая (N-ацетилнейраминовая) кислота узнавание клеток и их взаимодействие. Интересно, что углеводная часть структуры антигенов на поверхности эритроцитов (по системе АВО) может быть связана как с церамидом, так и с белками. В последнем случае структура антигена является не гликолипидом, а гликопротеином.
Некоторые ганглиозиды - рецепторы бактериальных токсинов. Например, GMl, находящийся на поверхности клеток кишечного эпителия, является местом прикрепления холерного токсина - белка, секретируемого возбудителями холеры.
Функции гликосфинголипидов можно суммировать следующим образом:
Взаимодействие между:
- клетками;
- клетками и межклеточным матриксом;
- клетками и микробами.
Модуляция:
- активности протеинкиназ;
- активности рецептора фактора роста;
- антипролиферативного действия (апоптоза, клеточного цикла).
Обеспечение:
- структурной жёсткости мембран;
- конформации белков мембран.
Синтез церамида и его производных. Синтез сфинголипидов начинается с образования церамида. Серии конденсируется с пальмитоил-КоА. Продукт их взаимодействия сначала восстанавливается коферментом NADPH, затем к аминогруппе дигидросфингозина амидной связью присоединяется жирная кислота, содержащая, как правило, 24 атома углерода. После окисления FAD-зависимой дегидрогеназой образуется церамид. Церамид служит предшественником в синтезе большой группы сфинголипидов: сфингомиелинов, не содержащих углеводов, и гликосфинголипидов. Последующие реакции синтеза катализируются специфическими трансферазами, набор которых отличается в разных тканях. Соединение фосфорилхолина с церамидом сфингомиелин-синтазой приводит к образованию сфингомие-лина. Присоединение углеводных компонентов катализируется специфическими гликозилтрансферазами. Донорами углеводных компонентов служат активированные сахара: УДФ-галактоза и УДФ-глюкоза. Галактоцереброзид - главный липид миелиновых оболочек; глюкоцереброзид входит в состав мембран многих клеток и служит предшественником в синтезе более сложных гликолипидов или продуктом на пути их катаболизма.
Катаболизм сфингомиелина. В лизосомах находятся ферменты, способные гидролизовать любые компоненты клеток. Эти ферменты называют кислыми гидролазами, так как они активны в кислой среде. Значение рН = 5, оптимальное для работы ферментов, создаётся протонным насосом, который, используя энергию АТФ, накачивает ионы водорода в лизосомы. Катаболизм сфингомиелинов и гликолипидов происходит в лизосомах. В распаде сфингомиелинов участвуют 2 фермента - сфингомиелиназа, отщепляющая фосфорилхолин, и церамидаза, продуктами действия которой являются сфингозин и жирная кислота
Катаболизм гликосфинголипидов. Катаболизм гликосфинголипидов начинается с перемещения их с поверхности клетки в цитоплазму по механизму эндоцитоза. В результате молекулы, расположенные на поверхности мембран, оказываются в эндоцитозных везикулах в цитоплазме и сливаются с лизосомами. В лизосомах находятся все ферменты, необходимые для гидролиза сложных молекул гликосфинголипидов: α- и β-галактозидазы, β-глюкозидазы, нейраминидаза (сиалидаза) и церамидаза. В результате последовательных реакций гидролиза сложные молекулы гликосфинголипидов распадаются до мономеров: глюкозы, галактозы, жирной кислоты, сфингозина и других метаболитов.
68.Нарушение обмена нейтрального жира (ожирение), фосфолипидов и гликолипидов. Сфинголипидозы
Ожирением считают состояние, когда масса тела превышает 20% от "идеальной" для данного индивидуума. Образование адипоцитов происходит ещё во внутриутробном состоянии, начиная с последнего триместра беременности, и заканчивается в препубертатный период. После этого жировые клетки могут увеличиваться в размерах при ожирении или уменьшаться при похудании, но их количество не изменяется в течение жизни.
Первичное ожирение. Первичное ожирение характеризуется множеством гормональных и метаболических особенностей у лиц, страдающих этим заболеванием. В самом общем виде можно сказать, что первичное ожирение развивается в результате алиментарного дисбаланса - избыточной калорийности питания по сравнению с расходами энергии. Суточные потребности организма в энергии складываются из:
- основного обмена - энергии, необходимой для поддержания жизни; основной обмен измеряют по поглощению кислорода или выделению тепла человеком в состоянии покоя утром, после 12-часового перерыва в еде;
- энергии, необходимой для физической активности.
Затраты энергии, необходимые для физической активности, разделяют на 3 уровня:
· I - 30% энергии от основного обмена (у людей, ведущих сидячий образ жизни);
· II - 60-70% от энергии основного обмена (у людей, которые 2 ч в день имеют умеренную физическую нагрузку);
· III - 100% и более от энергии основного обмена (у людей, которые в течение нескольких часов в день занимаются тяжёлой физической работой).
В зависимости от интенсивности нагрузки и возраста суточная потребность в энергии колеблется у женщин от 2000 до 3000 ккал в день, а у мужчин - от 2300 до 4000 ккал. Количество потребляемой пищи определяется многими факторами, в том числе и химическими регуляторами чувства голода и насыщения. Эти чувства определяются концентрацией в крови глюкозы и гормонов, которые инициируют чувство насыщения: холецистокинина, нейротензина, бомбезина, лептина.
Причины первичного ожирения:
- генетические нарушенвся (до 80% случаев ожирения - результат генетических нарушений);
- состав и количество потребляемой пищи, метод питания в семье;
- уровень физической активности;
- психологические факторы.
Генетические факторы в развитии ожирения. Метаболические различия между тучными и худыми людьми до настоящего времени не могут быть определены однозначно. Существует несколько теорий, объясняющих эти различия:
· генетически детерминированная разница в функционировании "бесполезных" циклов (субстратных циклов, раздел 7). Эти циклы состоят из пары метаболитов, превращаемых друг в друга с помощью двух ферментов. Одна из этих реакций идёт с затратой АТФ. Например:

· если эти субстраты превращаются друг в друга с одинаковой скоростью, то происходит "бесполезный" расход АТФ и, соответственно, источников энергии, например жиров;
· у людей, склонных к ожирению, вероятно, имеется более прочное сопряжение дыхания и окислительного фосфорилирования, т.е. более эффективный метаболизм;
· возможно, разное соотношение аэробного и анаэробного гликолиза. Анаэробный гликолиз (как менее эффективный) "сжигает" гораздо больше глюкозы, в результате снижается её переработка в жиры;
· у отдельных ивдивидуумов имеется различие в активности Nа+/К+-АТФ:азы, работа которой требует до 30% энергии, потребляемой клетками.
У человека и животных имеется " ген ожирения " - obese gene (ob). Продуктом экспрессии этого гена служит белок лептин, состоящий из 167 аминокислот, который синтезируется и секретируется адипоцитами и взаимодействует с рецепторами гипоталамуса. В результате его действия снижается секреция нейропептида Y. Нейропептид Y стимулирует пищевое поведение, поиск и потребление пищи у животных. Другие пептиды, участвующие в регуляции чувства сытости, например холецистокинин, также влияют на секрецию нейропептида Y. Таким опосредованным путём лептин выступает регулятором жировой массы, необходимой для роста и репродукции. Уровень лептина у больных ожирением может быть различным. У 80% больных концентрация лептина в крови тучных людей больше в 4 раза, чем у людей с нормальной массой тела. В этих случаях имеется генетический дефект рецепторов лептина в гипоталамусе, поэтому, несмотря на продукцию лептина, центр голода в гипоталамусе продолжает секрецию нейропептида Y. 20% больных имеют изменения в первичной структуре лептина. К настоящему времени описаны 5 одиночных мутаций в гене лептина, которые приводят к развитию ожирения. У этих больных наблюдают повышение отложения жиров в жировой ткани, чрезмерное потребление пищи, низкую физическую активность и развитие сахарного диабета типа II. Патогенез ожирения при дефекте гена ob может быть следующим: низкий уровень лептина в крови служит сигналом недостаточного количества запаса жиров в организме; этот сигнал включает механизмы, приводящие к увеличению аппетита и в результате к увеличению массы тела. Следовательно, можно сделать вывод о том, что первичное ожирение - не просто следствие переедания, а результат действия многих факторов, т.е. ожирение - полигенное заболевание.
| Ген ожирения |
| м-РНК |
| лептин |
| лептин + рецепторы гипоталамуса |
| Химические факторы (глюкоза, ХЦК и др.) |
| Центр голода |
| Нейропептид Y |
| Стимуляция пищевого поведения |
| Потребление пищи |
Генетический дефект сфингомиелиназы - причина болезни Ниманна-Пика. Дети с таким дефектом погибают в раннем возрасте. Симптомы заболевания: увеличение печени и селезёнки (гепатоспленомегалия), в лизосомах которых накапливается сфингомиелин; умственная отсталость. Генетический дефект другого фермента (церамидазы) приводит к развитию болезни Фарбера, симптомами которой также являются гепато- и спленомегалия, а также поражение суставов (болезненность, отёчность).
Генетические дефекты лизосомных ферментов катаболизма гликосфинголипидов. В норме синтез и катаболизм гликосфинголипидов сбалансированы таким образом, что количество этих компонентов в мембранах постоянно. Если имеется генетический дефект какого-либо лизосомного фермента, участвующего в катаболизме гликосфинголипида, то в лизосомах накапливается не-деполимеризованный субстрат, так называемые "остаточные тельца", размеры лизосом увеличиваются, их мембрана может разрушаться, ферменты выходят в цитозоль, и функции клеток нарушаются. Генетические заболевания вследствие дефекта какого-либо из ферментов катаболизма гликосфинголипидов называют сфинголипидоза-ми, или лизосомными болезнями. Эти заболевания редки, но среди некоторых популяций людей их частота очень высока. Так, болезнь Гоше вследствие дефекта фермента β-глюкрзидазы у евреев встречается с частотой 166:100 000, болезнь Тея-Сакса (дефект фермента β-гексозаминидазы) - с частотой 33:100 000. Сфинголипидозы обычно приводят к смерти в раннем возрасте, так как происходит поражение клеток нервной ткани, где сконцентрированы гликосфинголипиды. Однако при болезнях Гоше и Фабри больные живут, относительно долго.
Сфинголипиды, метаболизм: заболевания сфинголипидозы, таблица
| Заболевание | Фермент, недостаточностькоторого обусловливает заболевание | Накапливающийся:липид: | Клинические симптомы |
| Фукозидоз | альфа-Фукозидаза | Cer-Glc-GalNAc-Cal-:-Fuc Н-Изоантиген | Слабоумие, спастическое состояние мышц, утолщение кожи |
| Генерализованный ганглиозидоз | GM1-бета-Галактозидаза | Cer-Glc-Gal(NeuAc)-GalNAc-:-Gal Ганглиозид GM1 | Умственная отсталость, увеличениепечени, деформация скелета |
| Болезнь Тея-Сакса | Гексозаминидаза А | Cer-Glc-Gal-(NeuAc)-:-GalNAc Ганглиозид GM2 | Умственная отсталость, слепота, мышечная слабость |
| Вариант болезни Тея-Сакса, или болезнь Сандхоффа | Гексозаминидазы А и В | Cer-Glc-Gal-Gal-:-GalNAc Глобозид + ганглиозид GM2 | Те же, что и в случае болезни Тея-Сакса, но развиваются быстрее |
| Болезнь Фабри (сильно выражена только у мужчин); рецессивный генетический признак, связан с X-хромосомой | альфа-Галактозидаза | Cer-Glc-Gal-:-Gal Глоботриаозилцерамид | Кожная сыпь, почечная недостаточность |
| Церамидлактозидлипидоз | Церамидлактозидаза (бета-галактозидаза) | Cer-Glc-:-Gal Церамидлактозид | Прогрессирующее поражение мозга,увеличение печени и селезенки |
| Метахроматическая лейкодистрофия | Арилсульфатаза | Cer-Gal-:-OSO3 3-Сульфогалактозилцерамид | Умственная отсталость и психические нарушения у взрослых;демиелинизация |
| Болезнь Краббе | бета-Галактозидаза | Cer-:-Gal Галактозилцерамид | Умственная отсталость, почти полное отсутствие миелина |
| Болезнь Гоше | бета-Глюкозидаза | Cer-:-Glc Глюкозилцерамид | Увеличение печени и селезенки, эрозия трубчатых костей, умственная отсталость у детей |
| Болезнь Нимана-Пика | Сфингомиелиназа | Cer-:-P-холин Сфингомиелин | Увеличение печени и селезенки, умственная отсталость; фатальна в раннем возрасте |
| Болезнь Фарбера | Церамидаза | Ацил-:-Сфингозин Церамид | Хрипота, дерматит, деформация скелета, умственная отсталость; фатальна в раннем возрасте |
Обозначения: NeuAc - N-ацетилнейраминовая кислота; Cer - церамид; Glc - глюкоза; Gal - галактоза; Fuc - фукоза
69.Строение и биологические функции эйкозаноидов. Биосинтез простагландинов и лейкотриенов.
Эйкозаноиды, включающие в себя простагландины, тромбоксаны, лейкотриены и ряд других веществ, - высокоактивные регуляторы клеточных функций. Они имеют очень короткий Т1/2, поэтому оказывают эффекты как "гормоны местного действия", влияя на метаболизм продуцирующей их клетки по аугокзэинному механизму, и на окружающие клетки - по паракринному механизму. Эйкозаноиды участвуют во многих процессах: регулируют тонус ГМК и вследствие этого влияют на АД, состояние бронхов, кишечника, матки. Эйкозаноиды регулируют секрецию воды и натрия почками, влияют на образование тромбов. Разные типы эйкозаноидов участвуют в развитии воспалительного процесса, происходящего после повреждения тканей или инфекции. Такие признаки воспаления, как боль, отёк, лихорадка, в значительной мере обусловлены действием эйкозаноидов. Избыточная секреция эйкозаноидов приводит к ряду заболеваний, например бронхиальной астме и аллергическим реакциям.
Структура, номенклатура и биосинтез простагландинов и тромбоксанов Хотя субстраты для синтеза эйкозаноидов имеют довольно простую структуру (полистовые жирные кислоты), из них образуется большая и разнообразная группа веществ. Наиболее распространены в организме человека простагландины, которые впервые были выделены из предстательной железы, откуда и получили свое название. Позже было показано, что и другие ткани организма синтезируют простагландины и другие эйкозаноиды.
Не нашли, что искали? Воспользуйтесь поиском: