ТОР 5 статей:
Методические подходы к анализу финансового состояния предприятия
Проблема периодизации русской литературы ХХ века. Краткая характеристика второй половины ХХ века
Характеристика шлифовальных кругов и ее маркировка
Служебные части речи. Предлог. Союз. Частицы
КАТЕГОРИИ:
- Археология
- Архитектура
- Астрономия
- Аудит
- Биология
- Ботаника
- Бухгалтерский учёт
- Войное дело
- Генетика
- География
- Геология
- Дизайн
- Искусство
- История
- Кино
- Кулинария
- Культура
- Литература
- Математика
- Медицина
- Металлургия
- Мифология
- Музыка
- Психология
- Религия
- Спорт
- Строительство
- Техника
- Транспорт
- Туризм
- Усадьба
- Физика
- Фотография
- Химия
- Экология
- Электричество
- Электроника
- Энергетика
НЕКОТОРЫЕ СОПУТСТВУЮЩИЕ НЕСРАЩЕНИЯМ ВЕРХНЕЙ ГУБЫ И НЁБА СИНДРОМЫ ЧЕЛЮСТНО-ЛИЦЕВОЙ ОБЛАСТИ
Синдромные несращения губы и нёба составляют 10 % всех несращений лица. В настоящее время описано около 300 синдромов, связанных с пороками развития лица, челюстей и зубов. Из них, по последним данным, 70 % принадлежат к наследственным заболеваниям и около 30 % — к тератогенным порокам развития. Типы наследственности их изучены недостаточно.
Чаще всего встречаются такие синдромы: Пьера-Робина аномалад, Ван-дер-Вуда, Тричер-Коллинза, Апера, отопалатодигитальный, орофациальнодигиталь-ный синдром, EEC-синдром, синдромы несращения губ и нёба, сочетающиеся с эктодермальной дисплазией, с крыловидными складками на шее и в области суставов, с аномалиями больших пальцев кисти и микроцефалией; синдромы хромосомных аберраций, синдромы I—II жаберных дуг, микро- и макростомии и т. п. Очень редко наблюдаются врожденные несращения носа (рис. 341), "сиамские близнецы" с несращениями губы и нёба (рис. 342).

Рис. 341. Ребенок с врожденным несра- Рис. 342. "Сиамские близнецы", у одного
щением носа ("нос Тренделенбурга") из которых сквозное левостороннее несра-
щение верхней губы, альвеолярного отростка, твердого и мягкого нёба
•"irvn
Синдром Пьера-Робина аномалад. Тип наследования неизвестен. Врожденная гипоплазия нижней челюсти, микрогнатия, несращение нёба. Аномалии языка: глосоптоз, макроглоссия.
У 30 % таких детей диагностируют врожденные пороки сердца, аномалии глаз, ушных раковин, скелета и т. п. Нередко определяется умственная отсталость. После рождения ребенок плохо дышит в результате маленькой нижней челюсти и большого языка, который смещается кзади. Иногда язык приращен к мягким тканям дна ротовой полости. Во время сна возникают приступы асфиксии. Для нормализации дыхания ребенка кладут на живот, в тяжелых случаях язык в выдвинутом положении фиксируют к тканям полости рта. Прогноз благоприятный при адекватном раннем ортодонтическом лечении — изготовляют обтуратор, чтобы ребенок мог сосать. Кормление соской без обтуратора исключается. В тяжелых случаях на некоторое время применяют зондовое кормление. Используются и хирургические способы лечения — перемещение и фиксация языка в новом положении (кпереди от прежнего).
Синдром Ван-дер-Вуда. Наследуется по аутосомно-доминантному типу. Характерными являются врожденные симметричные свищи слизистых желез на нижней губе, сочетающиеся с несращением верхней губы (чаще всего с двусторонним сквозным). На медико-генетической консультации родителям следует сообщить о высоком (50 %) риске поражения потомков. Лечение хирургическое, от 3 мес до 1 года — хейлопластика, от 1 года до 2 лет — ураностафилоплас-тика, после этого — удаление свищей на нижней губе.
Синдром Тричер-Коллинза. Тип наследования неизвестен. Врожденный синдром с типичным комплексом симптомов: деформация или гипоплазия скуловых отростков лобных и височных костей, недоразвитие большей части скуловой дуги, нижние орбитальные края смещены книзу, орбиты деформированы, может отсутствовать наружный слуховой ход; деформация крыловидного отростка клиновидной кости, верхней и нижней челюстей, частичное несращение неба; нередко наблюдаются свищи на нижней губе, асимметрия и деформация черепа. Подбородок может быть недоразвитым, нос — деформированным.

|
Лечение хирургическое, многоэтапное.
Синдром Франческетти (черепно-че-люстно-лицевой дизостоз). Тип наследования аутосомно-доминантный. Комплекс врожденных челюстно-лицевых деформаций, сопровождающихся большим полуоткрытым ртом. Верхняя челюсть гипоплазирована, с уменьшенными придаточными пазухами. Высокое нёбо. Недоразвитая нижняя челюсть придает лицу птичий вид; возможны эпикантусы, антимонголоидный разрез глаз в резуль-
„»„ „ _ _ тате двусторонней гипоплазии скуловых
Рис. 343. Ребенок с синдромом Франчес- л
кетти (черепно-челюстно-лицевой дизо- костей и дуг. Определяется гипоплазия
стоз) обоих крыльев носа, что может ириво-
Раздел 10
Пороки развития тканей и органов челюстно-лицевой области
дить к сужению ноздрей. К основным признакам этого синдрома следует отнести отсутствие ресниц на нижних веках, наличие колобом, височно-предушное оволосение. Часто наблюдается атрезия наружного слухового прохода, полная глухота. В 50 % случаев имеет наследственный характер (рис. 343).
Лечение комплексное, длительное и многоэтапное.
Отопалатодигитальный синдром достаточно часто встречается в практике хирурга-стоматолога. Он проявляется глухотой, несращением нёба, дистрофиями лица и черепа (выступают лобные бугры и надбровные дуги, пшертелоризм, широкий нос с западением переносицы, микростома, микрогнатия). Также наблюдаются аномалии роста зубов, нарушения прикуса, мышечно-скелетные деформации, широкие и короткие ногтевые фаланги и отставание в умственном развитии (рис. 344).
Лечение комплексное, многоэтапное, длительное.
Орофациальнодигитальный синдром встречается в двух вариантах: 1-й — проявляется множественными уздечками языка и его частичным несращением, нес-рашением губы и нёба, несимметрично укороченными пальцами, аномалиями зубов, гипоплазией эмали. Также наблюдаются широкая спинка носа, аплазия крыльев носа и ушных раковин, эпнкантус и т. п.; 2-й вариант (синдром Мора) характеризуется гипертрофией уздечек, срединным псевдонесращением верхней губы, несращением нёба, отсутствием центральных резцов, гипоплазией скуловых дуг и челюстей, широкими переносицей и копчиком носа (рис. 345-348).
EEC-синдром (ectodactyli, ectodermal dysplasia and cleft palate s-m) характеризуется одно- или двусторонним несращением губы и нёба, редкими и тонкими волосами, сухой кожей, микродентией, изменением нормальной формы временных и постоянных зубов, гипоплазией эмали, стенозом слезных каналов. Также наблюдаются гипоплазия верхней челюсти и множественные пигментные неву-сы (рис. 349).
Лечение комплексное, многоэтапное.
Синдром гемифациальной микросомии (синдром I-II жаберных дуг). Тип наследования, повидимому, аутосомно-доминантный.
Это группа пороков, возникающих в результате нарушений формирования I жаберной щели, 1 и II жаберных дуг.
Характерными признаками являются: односторонняя микрогения, микрогнатия, недоразвитие скуловой кости и дуги, деформация наружного уха, атрофия и парезы мышц лица и нёба, языка, макростомия, околоушные свищи и придатки. Синдром I—Г Г жаберных дуг включает в себя и аномалии центральной нервной системы (олигофрению), деформации позвоночника, пороки развития мочеполовой системы, пищеварительного канала, врожденные пороки сердца, слепоту и т. п. Нередко диагностируются несращения верхней губы и нёба (рис. 350-353).
Лечение комплексное, длительное.
Синдром Апера (Apert) относится к сложным синдромам челюстно-лицевой области и скелета. Клиническими признаками его являются гипертелоризм, широкая переносица, плоские глазницы, пучеглазие, несращение нёба, слабое зрение. Наблюдаются изменения со стороны скелета — маленький рост, полидактилия. Этот синдром сопровождается умственной отсталостью (рис. 354).
Лечение, как, при предыдущих синдромах, многоэтапное, комплексное.
QO/1

Рис. 344. Изолированное врожденное несращение мягкого нёба и деформация пальцев рук (отопалатодигитальный синдром)

Рис. 346. Измененные фаланги пальцев ног у того же ребенка

Рис. 348. Врожденное двустороннее несращение верхней губы, альвеолярного отростка, твердого и мягкого нёба, сопровождающееся дефектами и деформациями верхних конечностей (орофациальнодигитальный синдром); ребенок после двусторонней хей-лопластики

Рис. 345. Ребенок с орофациальнодиги-тальным синдромом

Рис. 347. Синдактилия и шестипалость верхней конечности у того же ребенка
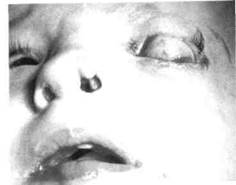
Рис. 349. Ребенок с врожденной левосторонней макростомией, дефектом и деформацией левого крыла носа и эктодермаль-ной дисплазией век (ЕЕС-синдром)
3Q5
Раздел 10
Пороки развития тканей и органов челюстно-лицевой области

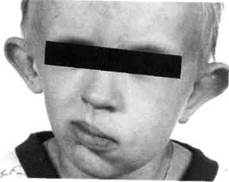
Рис. 350. Ребенок с синдромом Ml жаберных дуг (гемифациальная микросомия)

|
| "™ |
Рис. 352. Ребенок с синдромом гемифа-циальной микросомии (HI жаберных дуг)

Рис. 351. Ребенок с синдромом гемифа-циальной микросомии (I-II жаберных дуг)

Рис. 353. Ребенок с синдромом гемифа-циальной микросомии (I-II жаберных дуг) через 6 мес после хейлопластики

Синдром Клиппеля-Фейля (Klippel-Feil). В полости рта синдром проявляется нарушениями прикуса, несращениямп твердого и мягкого нёба, деформациями зубов, кистами челюстей. Внешний вид больного имеет типичный признак — короткую шею (голова "сидит" на плечах), движения головы ограничены. Это обусловлено тем, что I позвонок сливается с другими позвонками или синостози-рован с затылочной костью.
Кроме названных симптомов наблюдаются аномалии ребер, врожденные пороки сердца, высокое расположение лопаток.
Лечение многоэтапное и длительное, проводится соответствующими специалистами. В возрасте 1-2 года выполняется ураностафплопластика, если нет общесоматических противопоказаний, при выявлении кист челюстей — хирургическое лечение: кистотомия, кистэктомпя. Коррекцию прикуса и деформаций зубов осуществляет ортодонт.
Макро- и микростомии могут быть врожденными и сопровождать различные синдромы, определяющиеся в челюстно-лицевой области, и приобретенными, являющимися следствием ожогов, травм, хирургического вмешательства, проведенного лучевого лечения и т. п. Макро- и микростомия сопровождаются функциональными нарушениями (слюнотечением, выливанием пищи изо рта, ограниченным открыванием рта при микростомии, нарушением дикции) и различными косметическими деформациями.
Свыше 30 синдромов могут сопровождаться макро- и микростомиями: синдром Вольфа-Хиршхорна — хромосомы 4р (микростомия); Гольденхара синдром — скулоаурикуловертебральная дпсплазпя (макростомия); Казен синдром (микростомия); синдром Тричер-Коллинза (макростомия); синдром Франческетти (макростомия); Фримена-Шелдона синдром — краниокарпотарзальная диспла-зия (микростомия); Ханхарта синдром — синдром гипоглоссии-гиподактилии, синдром аглоссии-адактилии (микростомия); хромосомы 14р, 18д-синдром (микро- и макростомия соответственно) и т. п.
Лечебная тактика во всех случаях единственная — хирургическое вмешательство, предусматривающее при микростомии — расширение, а при макросто-мии — сужение ротовой щели. Существует много способов устранения этих деформаций: по А.И. Евдокимову, Г.О. Васильеву с использованием треугольных лоскутов, местной пластики по Ю.К. Шимановскому. В каждом конкретном случае в зависимости от степени нарушения функций, проявления косметических недостатков применяют ту или иную методику.
Колобома лица характеризуется несращением мягких тканей и костей по
трансверзали (поперечное) или сагитталп (косое), бывает одно- и двусторонней.
Сопровождается частичной гипоплазией тканей, атрофией мышц (рис. 355-358).
Лечение многоэтапное, длительное и заключается в анатомо-функциональ-
ном восстановлении несросшихся тканей (рис. 359, 360).
Рис. 354. Врожденная аплазия верхней челюсти
Рис. 355. Ребенок с правосторонней косой колобомой лица и отсутствием глазного яблока
газдел iu
 |

|

|
| Рис. 356.Ребенок с правосторонней коло-бомой лица |
| Рис. 358.Ребенок с правосторонней коло-бомой лица |

Рис. 357. Ребенок с двусторонней колобо-мой лица, несращением альвеолярного отростка, твердого и мягкого нёба

Рис. 359. Полость рта того же ребенка с правосторонней колобомой лица, атипичным несращением мягкого нёба и глотки

Рис. 360. Тот же ребенок через 6 лет после операции
Раздел 11
Основные
Не нашли, что искали? Воспользуйтесь поиском: