ТОР 5 статей:
Методические подходы к анализу финансового состояния предприятия
Проблема периодизации русской литературы ХХ века. Краткая характеристика второй половины ХХ века
Характеристика шлифовальных кругов и ее маркировка
Служебные части речи. Предлог. Союз. Частицы
КАТЕГОРИИ:
- Археология
- Архитектура
- Астрономия
- Аудит
- Биология
- Ботаника
- Бухгалтерский учёт
- Войное дело
- Генетика
- География
- Геология
- Дизайн
- Искусство
- История
- Кино
- Кулинария
- Культура
- Литература
- Математика
- Медицина
- Металлургия
- Мифология
- Музыка
- Психология
- Религия
- Спорт
- Строительство
- Техника
- Транспорт
- Туризм
- Усадьба
- Физика
- Фотография
- Химия
- Экология
- Электричество
- Электроника
- Энергетика
Титриметрические (объемные) методы
Наибольшее применение получил титриметрическии метод. Название происходит от слова "титр" (фр.) — концентрация. Основная операция метода—титрование, заключающаяся в постепенном приливании к раствору анализируемого вещества титрованного раствора до точки эквивалентности. По измеренному объему титрованного раствора рассчитывают количественное содержание вещества.
Титриметрическии метод анализа получил широкое распространение потому, что он позволяет использовать разнообразные химические реакции и определять вещества, учитывая их свойства и строение. Он выполняется быстро, с большой степенью точности, не нуждается в сложном оснащении и может использоваться как в лабораториях, так и в аптеках.
Для количественного определения лекарственного вещества титриметрическим методом необходимы титрованный (стандартный) раствор, набор простой лабораторной посуды (бюретки, пипетки, мерные колбы колбы для титрования) и средств фиксации точки эквивалентности (конечной точки титрования). Последнюю фиксируют как с помощью индикаторов, так и с помощью физико- химических методов, измеряя приборами физическую константу системы (потенциометрическое, амперометрическое титрование и др. способы). Однако не всякая химическая реакция может быть применима для процесса титрования. К реакциям, используемым в титриметрическом методе, предъявляются следующие требования:
1. возможность фиксировать точку эквивалентности (конечную точку титрования);
2. количественное протекание реакции, т. е. в реакцию должно вступить 100 % анализируемого вещества. Для этого необходимо строго соблюдать определенные условия титрования:
3. реакция должна протекать быстро;
4. не допускаются побочные реакции.
В зависимости от типа реакции, положенной в основу титрования, различают;
· кислотно-основное титрование;
· осадительное титрование;
· комплексиметрическое титрование;
· комплексонометрическое титрование;
· окислительно-восстановительное титрование.
Кислотно-основное титрование осуществляется в воде и в неводных средах. Данный метод используется в 40 процентах методик, применяющихся для анализа лекарственных веществ. Им определяют концентрацию кислот, оснований, солей. В основе титрования лежит реакция взаимодействия протонов с гидроксид-ионами: НзО+ + ОН- = 2Н2О. Титрованными (стандартными растворами являются растворы сильных кислот и сильных оснований. В процессе титрования изменяется рН системы. В зависимости от свойств определяемого вещества точка эквивалентности при титровании в воде может соответствовать различным величинам рН: Очевидно важно подобрать индикатор таким образом, чтобы величина рН в точке эквивалентности находилась в интервале перехода окраски выбранного индикатора.
В качестве индикаторов служат красители, изменяющие окраску в широком интервале рН от 1,2 до 10,5. Наиболее часто используются индикаторы: метиловый оранжевый (3,1—4,4); метиловый красный (4,8—6,0); фенолфталеин (8,2—10,0); тимол-фталеин (9,4—10,6).
Пример подбора индикатора. Подобрать индикатор для определения концентрации бензойной кислоты С6Н5СООН.
Решение. В процессе титрования бензойной кислоты (рКа=4,20) раствором гидроксида натрия образуется соль бензоат натрия
С6Н5СООН+NаОН = С6Н5СOONа+Н20
Бензоат натрия в воде подвергается процессам диссоциации и гидролиза:
С6Н5СООNа -> С6Н5СОО- + Nа+;
С6Н5СОО- + Н20 = С6Н5СООН + ОН-.
В растворе накапливается определенное количество ОН-, их концентрация превысит концентрацию протонов, и поэтому величина рН будет более 7. Это подтверждается приведенными ниже расчетами по формуле рН для растворов солей слабых кислот и сильных оснований: рН = 7 + ½*рКа + ½*lgСсоли, где рКа = 4,20 (табличная величина), а Ссоли определяют, ориентируясь на концентрацию титрованного раствора. Если титруют 0,1 М раствором гидроксида натрия, то Ссоли = 0,1 моль/л. В этом случае lgС = lg0,1 и lgС = -1; ½*0,1 = -0,5. Подставив значения рКа и ½*lgС в рН = 7 + ½*4,20— 0,5=8,6, найдем значение рН в точке эквивалентности. Эта величина находится в интервале рН для фенолфталеина (8,2 — 10,0), следовательно, бензойную кислоту нужно титровать с индикатором фенолфталеином.
Значительное количество лекарственных веществ проявляет способность отщеплять или присоединять протоны и согласно современным теориям являться кислотами или основаниями. Мерой кислотности вещества служит величина показателя кислотности рКа = -1gКа, где Ка— константа ионизации. Чем меньше величина рКа, тем сильнее кислота, тем легче отщепляются протоны. Аналогично рКв — показатель основности вещества. Чем меньше величина рКв, тем сильнее основание, тем активнее вещество присоединяет протоны. Значения рКа и рКв для одного и того же вещества в разных растворителях различны, и этот фактор используют для выбора условий титрования.
ГФ XI приводит значения рКа для ряда лекарственных веществ в различных растворителях. Зная величину рКа, можно решить вопрос о возможности и условиях титрования вещества. Например, для соляной кислоты в воде рКа=0,8; для уксусной кислоты рКа=4,75; для ацетилсалициловой кислоты рКа=3,50. Эти кислоты можно титровать в воде раствором гидроксида натрия. Если величина рКа больше восьми единиц рН, то водная среда не подходит. Например, для титрования барбитала (рКа=7,47), фенола (рКа=9,89), борной кислоты (рКа=9,24) требуются особые условия. Барбитал титруют в среде диметилформамида бензольно-метанольным раствором гидроксида натрия. Борную кислоту превращают добавлением глицерина в диглицеринборную кислоту, которая является более сильной кислотой.
Свои основные свойства в водных и спиртовых средах проявляют лекарственные вещества, присоединяющие протон. Это—амидопирин (рКв=9,2), гексаметилентетрамин (рКв=9,1), алкалоиды, например кодеин (рКв=6,0),. поэтому их можно титровать раствором сильной кислоты.
В водной среде кислотами титруют натриевые соли слабых кислот, так как в их растворе вследствие гидролиза образуется щелочная среда. Соли алкалоидов, в водных растворах которых возникает кислая среда вследствие гидролиза, титруют раствором гидроксида натрия. В процессе титрования солей образуются кислоты или основания, присутствие их оказывает существенное влияние на рН раствора, поэтому их удаляют путем экстрагирования растворителями, не смешивающимися с водой. Например, салицилат натрия, бензоат натрия титруют в присутствии эфира. А соли алкалоидов в присутствии спиртово-хлороформной смеси (1:1). Для алкалиметрического определения аминокислот используется метод формольного титрования (титрование по Серенсену). Наличие аминогруппы, способной присоединять протоны, и карбоксильной группы, отдающей протоны, приводит к тому, что в водных растворах аминокислоты существуют в виде диполярных ионов +NH3-R-СОО-, поэтому полностью оттитровать такие вещества раствором гидроксида натрия не удается. Во избежание этого в раствор перед титрованием добавляют нейтрализованный формалин. Образуется ТЧ-метиленовое производное и устраняется влияние аминогруппы

Если вещество — очень слабая кислота с рКа > 9, например теофиллин (рКа=11,40), его непосредственно оттитровать нельзя. В таком случае прибегают к заместительному титрованию, сущность которого заключается в том, что к раствору анализируемого вещества добавляют несколько капель раствора нитрата серебра. Выделяющееся эквивалентное количество азотной кислоты определяют, алкалиметрически:

Титрование в неводных средах имеет преимущество перед водным титрованием потому, что позволяет определять концентрацию слабых кислот и оснований, часто мало растворимых в воде. Этот метод позволяет также определять соли слабых кислот и слабых оснований, которые невозможно оттитровать в воде. Удобен метод и для анализа многокомпонентных смесей,, часто без их предварительного разделения. Метод позволяет определять физиологически активную часть в солях алкалоидов.
Метод неводного титрования дает более точные результаты по сравнению с титрованием в воде, так как вследствие небольшого поверхностного натяжения неводных растворителей размеры капель титрованных растворов меньше капель водных растворов.
В теории неводного титрования большую роль играет влияние растворителя, на кислотно-основные свойства анализируемого вещества. Для неводного титрования применяются различные растворители, которые по своим свойствам делятся на четыре группы.
Основные (протофильные) растворители легко присоединяют протоны, усиливают кислотные свойства титруемых веществ. Среди них — диметилформамид НСОN(СН3)2, пиридин, жидкий аммиак и др. В среде основных растворителей легко титруются слабые кислоты, кислые формы.барбитуратов, сульфаниламидов, фенолы. Кислотность этих соединений в среде данных растворителей повышается, и тем самым улучшается процесс и результаты титрования. Титрантом служит раствор гидроксида натрия в смеси метанола и бензола или раствор метилата натрия. В качестве индикатора применяют тимоловый синий. Например, при титровании барбитала в среде диметилформамида раствором гидроксида натрия происходят следующие процессы:

2. Кислотные (протогенные) растворители: муравьиная кислота НСООН, уксусная кислота СН3СООН (безводная), уксусный ангидрид и др. Они легко отдают протоны, усиливая основные свойства веществ. Титрантом служит раствор хлорной кислоты, а индикатором — раствор кристаллического фиолетового, тропеолина 00 или метилового оранжевого. Растворы титранта и индикатора готовят в безводной уксусной кислоте. Суммарно процесс нейтрализации слабого органического основания хлорной кислотой представлен следующей схемой:
R3N + HClO4 → [R2N • H+]ClO4-
Подобно происходит титрование производные пиридина (никотинамид, фтивазид), алкалоидов, представляющих собой слабые основания.
Амфотерные (амфипротные) растворители: вода Н2О, этанол С2Н5ОН, метанол СН3ОН и др. Эти растворители могут отдавать свои или присоединять протоны от титруемых веществ. В амфипротных растворителях титруют смеси различных кислот.
Индифферентные (апротонные) растворители: углеводороды — бензол и его производные, галоген-производные углеводородов (хлороформ, четыреххлористый углерод и др.). Молекулы этих растворителей не способны ни отдавать, ни присоединять протоны. В них титруются смеси оснований.
Недостатком неводного титрования является необходимость иметь герметизированную титровальную установку. Работа предполагает использование токсичных, летучих растворителей. Однако метод позволяет определять концентрацию солей слабых кислот и слабых оснований, что не всегда возможно в водной среде. Ацетат калия титруется хлорной кислотой по схеме
СН3СООК + HClO4 →KClO4 + СН3СООН.
Титрование солей слабых оснований (R3N • HA) •можно выразить следующей схемой
R3N • HA + HClO4 → [R3N • H]ClO4 + HA.
Так титруются адреналин и норадреналин гидротартраты, нафтизин, цитрат дитразина, соли алкалоидов (фосфат кодеина, гидротартрат платифиллина, сульфат атропина, бензоат сферофизина). Однако соли галогенводородных кислот (гидрохлориды, гидробромиды, гидроиодиды) алкалоидов и азотсодержащих оснований не могут быть непосредственно оттитрованы хлорной кислотой, так как галоген-ионы проявляют кислотные свойства даже в среде безводной уксусной кислоты и поэтому могут влиять на переход цвета индикатора в точке эквивалентности. Титрование солей галогенводородных кислот выполняют в присутствии ацетата ртути (II), который связывает галоген-ионы в малодиссоциированные соединения (дихлорид, дибромид или дийодид ртути), и титрование идет с хорошими результатами по схеме
2R3N • HX + Hg(CH3COO)2 → HgX2 + 2[R3NH]+CH3COO-.
2[R3NH]+CH3COO- + 2HClO4 → 2[R3NH]+ClO4 + 2CH3COOH.
Возможность и оптимальные условия титрования в неводных средах определяются величиной константы титрования Кт, которую рассчитывают, исходя из величин ионного произведения среды Кi и Ка — константы диссоциации титруемого вещества в этой среде, по формулам для кислот Кт = Кi/Ка, для оснований Кт=Ка или рКт=рК; -рКа и рКт=рКа. Чем меньше числовое значение Кт и чем больше рКт, тем условия титрования лучше. Значения величины Кi; и рКi; для ряда растворителей и Ка, а также рКа для некоторых лекарственных веществ приведены в ГФ XI.
Пример выбора среды для количественного определения ацетилсалициловой кислоты.
Величина рКi этанола 19,1; воды — 14. Для ацетил-салициловой кислоты рКа=3,50. В воде рКт=14-3,50=10,5; в этаноле рКт=19,1-3,50=15,6.
Величина рКт в этаноле больше, следовательно в этом растворителе условия титрования ацетилсалициловой кислоты лучше.
В ряде случаев для титрования применяют смеси неводных растворителей с апротонными растворителями: бензолом, хлороформом и др., присутствие которых уменьшает ионное произведение среды К; что способствует улучшению условий титрования.
Осадительное титрование. В основу метода положе на реакция образования малорастворимого соединения, В фармацевтическом анализе широко используют аргентометрию, которая предполагает взаимодействие галогенов с нитратом серебра:
МеНаl + АgNО3 → АgНаl↓ + МеNO3.
Применяется метод в виде прямого (методы Мора, Фаянса) и обратного титрования (метод Фольгарда) Титрантами являются 0,1 М и 0,05 М растворы нитрат серебра и тиоцианата аммония.
По методу Мора титрование раствором нитрата серебра выполняют при рН 6,5—10,0 в присутствие 5—7 капель 5 %-ного водного раствора хромата калия в качестве индикатора. В процессе титрования образуются малорастворимые галогениды серебра, и, когда и осаждение закончится полностью, образуется красный осадок хромата серебра, свидетельствующий о достижении точки эквивалентности:
К2СrО4, + 2АgNO3 → Ag2СrО4↓ + 2КNO3
Этим методом определяют концентрацию хлоридов и бромидов. Иодиды определять не рекомендуется, потому что появление красной окраски происходит ранее точки эквивалентности, что объясняется адсорбцией иодид-ионов поверхностью осадка.
Метод Фаянса применяется для определения концентрации йодидов, но он может использоваться также для хлоридов и бромидов. В отличие от метода Мора, титрование выполняется не только в нейтральной среде, но и в среде уксусной кислоты с водным раствором эозината натрия в качестве индикатора. В точке эквивалентности наблюдается появление ярко-розового окрашинивания осадка. Хлориды и бромиды титруют в среде уксусной кислоты, индикатором служит раствор бром-фенолового синего. В точке эквивалентности зеленовато-желтое окрашивание переходит в сине- фиолетовое. Метод Фольгарда используется для определения концентрации хлоридов, бромидов, йодидов способом обратного титрования. Индикатором является раствор железоаммониевых квасцов. Анализ производится в среде азотной кислоты.К отобранному для определения концентрации раствору приливают точно измеренный избыток раствора нитрата серебра, 2—3 мл разведенной азотной кислоты, 10 капель раствора железоаммониевых квасцов и титруют раствором тиоцианата аммония до появления розовой окраски:АgNО3 + NН4SCN → AgSСN↓ + NH4NO3,
3NН4SCN + FeNH4(SO4)2 → [Fe(SCN)3] + 2(NH4)2SO4
При титровании хлоридов возможно взаимодействие осадка хлорида серебра с красным соединением:
[Fe(SCN)3] + 3АgСl → 3АgSСN↓ + FеС13
и определение точки эквивалентности затрудняется. Чтобы избежать протекания реакции между хлоридом серебра и комплексным соединением, можно отфильтровать осадок и в фильтрате оттитровать избыток нитрата серебра. Но можно перед титрованием в анализируемый раствор добавить 5—10 мл органического растворителя с большой плотностью, например четыреххлористого углерода, который покрывает поверхность осадка хлорида серебра, и тогда взаимодействие осадка не происходит. При титровании йодидов индикатор — раствор железоаммониевых квасцов — прибавляют после добавления избытка нитрата серебра. Если этого не сделать, то возможно окислительно-восстановительное взаимодействие йодид-иона с индикатором
2КI + 2 FeNH4(SO4)2 → 2FeSO4 + I2 + (NН4)2SO4 + К2SO4.
Видоизмененный метод Фольгарда используется при определении хлоридов и йодидов. Этот способ позволяет избежать взаимодействия йодидов с железоаммониевыми квасцами и осадка хлорида серебра с комплексным соединением [Fe(SCN)3] и тем самым улучшить условия титрования.
К растворенной навеске галогенида добавляют 2—3 мл разведенной азотной кислоты, 1 мл раствора железоаммониевых квасцов и 0,1 мл 0,1 М раствора тиоцианата аммония. Раствор окрашивается в красный цвет. Титруют 0,1 М раствором нитрата серебра до исчезновения окраски. При расчетах необходимо учитывать объем раствора нитрата серебра, который затратится на 0,1 мл 0,1 М раствора тиоцианата аммония, добавленного в анализируемый раствор до начала титрования.
Описанные выше методы осадительного титрования не являются избирательными, при анализе раствора смеси галогенидов определяется их общее содержание. Для определения йодидов в растворах с хлоридами и бромидами существуют избирательные методы.
Метод Кольтгофа является методом прямого аргентометрического титрования раствором нитрата серебра. К анализируемому раствору приливают 3 мл воды, 3 мл 10%-ного раствора карбоната аммония, 3—4 капли разведенной серной кислоты, 5—6 капель раствора крахмала, одну каплю 0,1 М раствора йодата калия. Раствор окрашивается в синий цвет вследствие выделения йода согласно уравнению
5KI + КIO3 + 3H2SO4 → 3I2 + 3K2SO4 + 3Н20.
Раствор медленно, тщательно перемешивая, титруют раствором нитрата серебра до перехода синей окраски в желтую, обусловленную цветом осадка йодида серебра. В процессе титрования в растворе уменьшается концентрация йодида калия, равновесие смещается влево, уменьшается концентрация йода, и синяя окраска исчезает Возможность определения йодидов в присутствии остальных галогенидов достигается потому, что в растворе образуется буферная смесь, поддерживающая значение рН<5,5. Бромиды в этих условиях не окисляются йодатом калия при его незначительной концентрации.
Другим методом прямого аргентометрического титрования является метод Шика. К анализируемому раствору приливают 4—5 мл воды, 5 мл разведенной серной кислоты и титруют раствором нитрата серебра. Индикатор — нитрозокрахмальная бумага (полоска фильтровальной бумаги, пропитанная смесью растворов нитрита натрия и крахмала). При нанесении на ее поверхность капли раствора до момента эквивалентности бумага окрашивается в синий цвет вследствие протекания реакции
2NaNO2 + 2NaI + 2H2SO4 → I2 + 2Na2SO4 + 2H20 + 2NO
Цвет бумаги не изменится после достижения точки эквивалентности. Для получения более точных результатов целесообразно предварительно рассчитать количество титрованного раствора нитрата серебра, необходимого для титрования взятого количества йодидов, или выполнить ориентировочное титрование, а затем при повторном титровании иметь в виду результаты расчетов.
Несколько видоизмененный метод Фольгарда нашел применение не только для определения содержания галогенид-ионов. В его основе лежит способность некоторых органических веществ осаждаться солями серебра.
Например, концентрацию никотиновой кислоты можно определить, после нейтрализации ее аликвотной части, осаждением точно измеренным избытком титрованного раствора нитрата серебра. Происходит образование малорастворимой соли. Через 30 мин осадок отфильтровывают. К аликвотной части фильтрата прибавляют несколько капель разведенной азотной кислоты, раствор железоаммониевых квасцов и титруют 0,1 М раствором тиоцианата аммония до появления розового окрашивания раствора.
Концентрацию дибазола выявляют следующим образом: вначале его осаждают в виде малорастворимой соли серебра, затем осадок с фильтром переносят в колбу для титрования, прибавляют 1—2 мл разведенной азотной кислоты, слегка подогревают до растворения осадка. Охлажденный раствор титруют раствором тиоцианата аммония, используя в качестве индикатора железоаммониевые квасцы до появления розовой окраски раствора.
Для определения содержания сульфатов титрантами служат 0,1 М растворы солей нитрата свинца и нитрата бария. С целью создания оптимальных условий титрования в анализируемый раствор добавляют ацетон или 95%-ный спирт.
Сульфат атропина определяют титрованием раствором нитрата сйинца с индикатором дифенилкарбазидом до перехода желтого окрашивания в розовое. Концентрацию некоторых сульфатов можно выявлять титрованием раствором нитрата бария в присутствии в качестве индикатора 0,2%-ного водного раствора карбоксиарсеназа. Так определяют сульфат атропина, сульфат стрептомицина, сульфат цинка, сульфат хинина.
Комплексиметрический метод основан на образовании комплексного соединения. Меркуриметрия используется для определения концентрации галогенидов, тиоцианатов, цианидов с помощью титрованного раствора — нитрата ртути (II). Предложен также раствор перхлората.ртути (II). Титрованные растворы готовят с добавлением соответствующих кислот. Точку эквивалентности устанавливают потенциометрически или с помощью индикатора дифенилкарбазида, образующего с избытком соли ртути (II) сине-фиолетовое соединение:
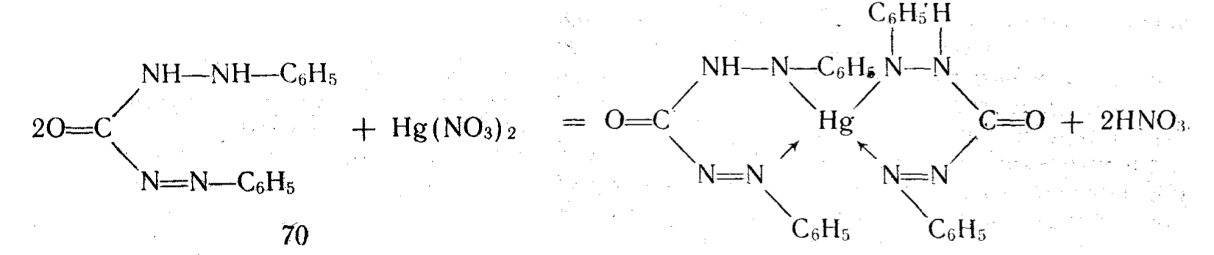
При определении йодидов в процессе титрования образуется бесцветное комплексное соединение 4КI + Hg(NO3)2= К2[НgI4] + 2КNO3.
Точку эквивалентности определяют по образованию неисчезающего красного осадка дийодида ртути (II) К2[HgI4] + Нg(NO3)2 = 2HgI2 + 2КNО3.
Иодиды можно титровать с индикатором дифенилкарбазидом, если в титруемый раствор добавить несколько миллилитров этанола. Красный осадок дийодида ртути (II) растворяется в этаноле, и тогда точку эквивалентности определяют с индикатором по появлению сине-фиолетового окрашивания. Титрование выполняется в кислой среде.
При работе с солями ртути (II) необходимо соблюдать осторожность, помнить, что растворимые соли ртути ядовиты.
К комплексиметрическому титрованию относится куприметрическое определение левомицетина. К раствору левомицетина добавляют несколько капель раствора гидроксида натрия, мурексид (как индикатор) и медленно приливают титрованный раствор сульфата меди до изменения окраски раствора из фиолетовой в коричнево-красную, сравнивая ее с окраской контрольного раствора.
При добавлении гидроксида натрия к раствору левомицетина происходит взаимодействие

В процессе титрования образуется комплексное соединение:

Комплексонометрический метод основан на реакции образования прочных комплексов полиаминокарбоновых кислот с ионами металлов (Са2+, Mg2+,Zn2+, Вi2+ и др.). Наиболее широко применяется динатриевая соль этилендиаминтетрауксусной кислоты (трилон Б). Трилон Б наряду с карбоксильными группами содержит аминный азот. Вследствие такого строения он является кислотой и комплексообразующим веществом. Многие металлы заменяют атомы водорода карбоксильных групп, одновременно связываясь координационно с азотом аминогруппы и образуя прочные комплексы трилона Б с металлом. Двухзарядный катион (например, Са2+) образует комплексное соединение следующего состава:
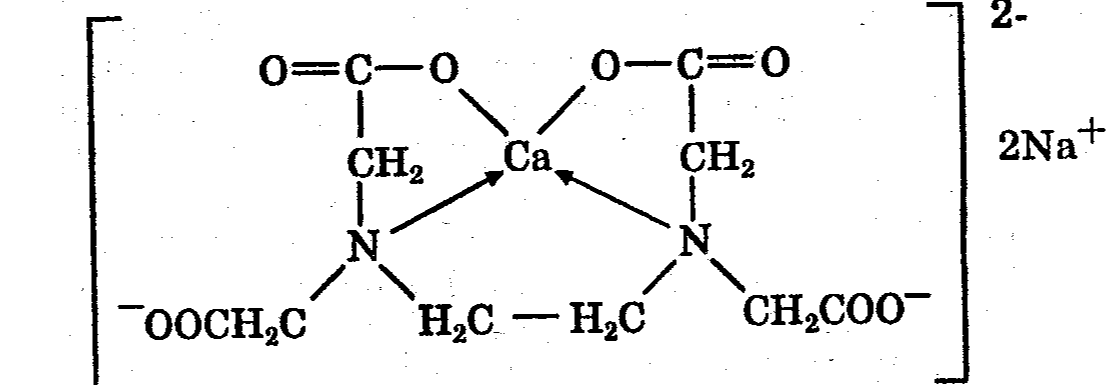
Образование комплексов можно представить схематично:
Na2H2I → 2Na+ + H2I2-
Ме2+ + H2I2- → MeI2- + 2Н+
где Na2H2I — трилон Б; Ме2+ — ион металла. Как видно из приведенной схемы, реакция образования комплексов сопровождается накоплением протонов в растворе, поэтому связывание Н+- ионов должно способствовать образованию комплекса. Наиболее благоприятный для комплексообразования реакцией среды является рН 8—10. Поэтому титрование солей металлов трилоном Б проводят в присутствии аммиачного буфера. Для установления точки эквивалентности применяются специальные индикаторы, которые являются органическими красителями. К ним относятся кислотный хром темно-синий, кислотный хром черный специальный, называемый эриохром черный Т, мурексид, калькон-карбоновая кислота и др. Процесс комплексонометрического титрования заключается в том, что к исследуемому раствору, содержащему определяемый катион, при строго определенном значении рН прибавляют индикатор, при этом образуется хорошо растворимое в воде окрашенное комплексное соединение индикатора с ионом определяемого металла. При титровании трилоном Б этот комплекс разрушается и образуется более прочный, как правило бесцветный, комплекс иона металла с трилоном Б. При этом выделяется анион индикатора, который окрашивает раствор в цвет, присущий свободному индикатору при данном значении рН:
Са2+ + Н2Ind- → CaInd- + 2Н+
CaInd- + H2I2- → CaI2- + H2Ind-
Комплексонометрическое титрование осуществляется как методом прямого, так и методом обратного титрования. Оно позволяет определять количественное содержание солей, оксидов металлов магния, кальция, цинка, свинца, висмута, ртути и др. Метод пригоден также для раздельного определения солей металлов в смеси. Раздельное определение солей кальция и магния при их совместном присутствии основано на том, что растворимость соединений титруемых солей зависит от величины рН в анализируемом растворе. Аликвотную часть раствора титруют вначале с индикатором эриохром черным при рН = 9 в присутствии аммиачного буферного раствора, причем титруются обе соли. В другой аликвотной части определяют соль кальция. В раствор добавляют несколько миллилитров 20%-ного раствора гидроксида натрия, рН этого раствора изменяется от 9 до 12. В этих условиях соли магния осаждаются в виде гидроксида магния, и далее титруют соль кальция с индикатором мурексидом.
Интерес представляет косвенное комплексонометрическое определение аминопроизводных и солей органических оснований (гидрохлорида папаверина, прозерина, спазмолитина, производных фенотиазина). В этом случае используется раствор тетрароданоцинкат (П)-аммония (ТРЦ), который получают при взаимодействии тиоцианата аммония и сульфата цинка:
Для приготовления 0,5 М раствора ТРЦ берут 144 г сульфата цинка, помещают в мерную колбу вместимостью 1 л, добавляют 152 г тиоцианата аммония, растворяют в воде и доводят водой до 1 л. Тщательно перемешивают, фильтруют через вату, хранят при комнатной температуре. При хранении реактива может выпасть осадок или измениться цвет. Несмотря на эти изменения, реактив годен к применению.
4NH4SCN + ZnSO4 → (NH4)2[Zn(SCN)4] + (NH4)2SO4
При добавлении реактива ТРЦ к анализируемому раствору образуется осадок комплексной соли. Например, при определении прозерина происходит осаждение его в соответствии с уравнением реакции

Осадок экстрагируют точно измеренным объемом хлороформа при энергичном взбалтывании в течение 2 мини фильтруют через бумажный фильтр в сухую колбу. В колбу для титрования отбирают пипеткой определенный объем фильтрата, приливают избыток титрованного раствора трилона Б. Часть титранта вступает во взаимодействие с цинком, образуя прочное комплексное соединение. Не вошедший в реакцию титрант в присутствии аммиачного буферного раствора и индикатора хром темно-синего оттитровывают раствором сульфата цинка.
Окислительно-восстановительное титрование основано на окислительных или восстановительных свойствах анализируемых веществ и титрантов. В процессе титрования происходит изменение окислительно-восстановительных потенциалов взаимодействующих систем. Если разность этих потенциалов достаточно большая, то окислительно-восстановительный процесс протекает практически до конца, и поэтому возможно прямое титрование.
В фармацевтическом анализе применяют такие методы окислительно-восстановительного титрования, как перманганатометрия, йодометрия, йодхлорометрия, йодатометрия, броматометрия, дихроматометрия, цериметрия.
ПЕРМАНГАНАТОМЕТРИЯ основана на использовании окислительных свойств титранта — перманганата калия в кислой и щелочной средах.
МnО4- + 8Н+ + 5е → Мn2+ + 4Н20.
В кислой среде продуктом восстановления являются практически бесцветные соли марганца (II). Поэтому при прямом перманганатометрическом титровании индикатор в анализируемый раствор не добавляют, им является титрант, избыток которого придает раствору розовое окрашивание.
Прямое титрование используется для определения восстановленного железа, препаратов пероксида водорода.
В случае медленного протекания окислительно-восстановительного процесса применяется обратное титрование. Например, при определении нитрита натрия, левомицетина. Избыток титранта оттитровывают йодометрически.
ИОДОМЕТРИЯ основана на использовании окислительных свойств свободного йода и восстановительных свойств йодид-ионов:
I2 + 2е → 2I-
Методом йодометрии количественно определяют неорганические и органические лекарственные вещества, способные окисляться или восстанавливаться, а также образовывать с йодом продукты замещения. Кроме того, йодометрию используют для определения избытка титранта в обратном окислительно-восстановительном титровании. Свободный йод, образовавшийся в избытке при обратном йодометрическом титровании, оттитровывают тиосульфатом натрия:
I2 + 2Nа2S2О3 → 2NаI + Nа2S4О6.
Индикатором обычно служит раствор крахмала, образующий с йодом синее соединение. Прямое титрование йодом применяют для определения тиосульфата натрия и препаратов мышьяка (III). Иодометрическое опреде ление, основанное на окислении альдегидов йодом, используют для титрования хлоралгидрата, формальдегида в растворе, а также лекарственных веществ образующих формальдегид при гидролизе (никодина метазида). Окисление альдегидов происходит по схеме:
I2 + 2NаОН → NaIO + NaI + Н2O
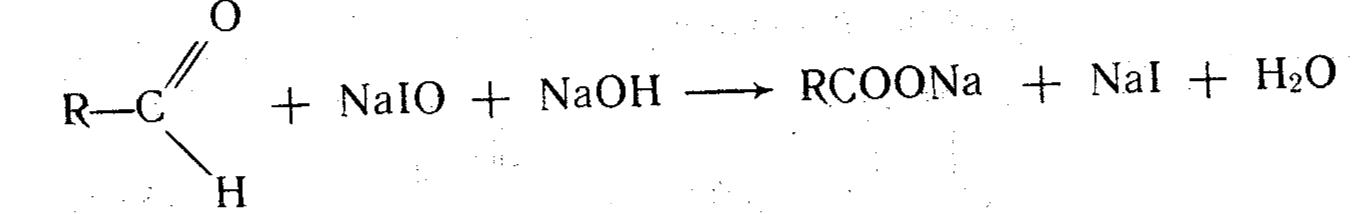
Процесс окисления йодом лежит в основе определении фурацилина, изониазида, метионина, анальгина и др.
Восстановительные свойства йодида калия используются для определения окислителей. Лекарственные вещества — окислители выделяют эквивалентное количество свободного йода при взаимодействии с йодидом калия. Выделившийся йод титруют раствором тиосульфата натрия. Эти процессы лежат в основе колчественного определения препаратов пероксида водорода, соединений мышьяка (V), меди (II), перманганата калия, а также обладающих окислительными свойствами гипохлоридов (известь хлорная) и хлор-производных амидов сульфокислот (хлорамины, пантоцид). Для количественного анализа используется сочетание реакций замещения и йодометрии. С помощью титрованного раствора йода получают йодпроизводные, их отфильтровывают, а в фильтрате определяют избыток йода титрованием раствором тиосульфата натрия. Этот прием используют для определения некоторых алкалоидов, которые образуют малорастворимые перйодиды состава [R3N]•НI•I4. Это кодеин, кокаин, кофеин и др.
ИОДХЛОРОМЕТРИЮ согласно ГФ XI рекомендуют для количественного определения лактата этакридина, который осаждается в виде дийодпроизводного. Избыток титранта раствора йодмонохлорида оттитровывают йодометрически:
IСl + КI →I2 + КСl.
Иодхлорометрическим методом можно определять фенолы, сульфаниламиды и другие первичные ароматические амины.
ИОДАТОМЕТРИЮ используют для определения фтивазида, апрессина, аскорбиновой кислоты. Происходит процесс окисления лекарственных веществ титрованным раствором йодата калия КIO3. Например, по ГФ X аскорбиновую кислоту рекомендуют титровать 0,1 М раствором йодата калия в присутствии йодида калия. Окисление происходит по схеме:

Избыток титранта йодата калия в точке эквивалентности приводит к окислению йодида калия в кислой среде в соответствии с уравнением
КIO3 + 5KI + 6НСl → 3I2 + 6KCl + 3Н20.
Иод окрашивает раствор в желтый цвет, а после добавления раствора крахмала — в синий цвет. Фтивазид титруют раствором йодата калия после предварительного гидролиза в среде соляной кислоты. В титруемый раствор прибавляют несколько миллилитров хлороформа. Образующийся при титровании йод извлекается хлороформом, окрашивая его в фиолетовый цвет: Точку эквивалентности определяют по обесцвечиванию хлороформного слоя, когда йод превратится в монохлорид иода.
В БРОМАТОМЕТРИИ в качестве титранта применяется раствор бромата калия. Титрование восстановителей, таких, как мышьяк (III), сурьма (III), гидроксиламин, производные гидразина и др., можно осуществлять прямым и обратным титрованием в среде соляной и серной кислот. Бромат калия восстанавливается до бромида калия, и в тот момент, когда в растворе появляется небольшой избыток титранта, тотчас реагирует с ним:
КBrО3 + 5KBr + 6НС1 → 3Br2 + 6КС1 + 3Н20.
Образовавшийся свободный бром окрашивает раствор в бледно-желтый цвет. Однако эта окраска очень слабая и точку эквивалентности по ней точно фиксировать нельзя, поэтому пользуются индикаторами метиловым оранжевым и метиловым красным. В точке эквивалентности индикатор необратимо окисляется до бесцветных продуктов.
Броматометрические определения основаны не только на окислительно-восстановительных процессах, но и на реакциях присоединения брома и замещения бромом, который образуется в процессе взаимодействия с бромидом калия. Поэтому очень часто титрование производят титрованным раствором, содержащим бромат калия и бромид калия в соотношении 1:5. При использовании метода обратного титрования избыток титранта определяют йодометрически. Метод используют для определения лекарственных веществ производных фенолов (фенол, тимол, резорцин, салициловая кислота) и первичных ароматических аминов (сульфаниламидные препараты, производные п-аминобензойной кислоты), йодидов и органических оснований.
ДИХРОМАТОМЕТРИЯ использует в качестве титранта раствор дихромата калия. Титрование выполняется в среде серной, соляной или фосфорной кислот.
Применение этого метода основано на реакциях окисления-восстановления и реакциях двойного обмена, сопровождающихся образованием не растворимых в воде соединений. Так определяют концентрацию метиленового синего, акрихина, этония. К растворенной навеске анализируемого вещества приливают избыток титрованного раствора, происходит образование осадка дихромата основания:
2 (R3N • Н)+Сl- + К2Сr2О7 → [R3N • Н]2Сг2O7↓ + 2КС1.
После отделения осадка фильтрованием избыток титранта в фильтрате определяют йодометрическим методом:
К2Сr2О7 + 6KI + 7H2SO4 → Cr2(SO4)3 + 3I2 + 4K2SO4 + 7Н20.
Выделившийся йод титруют раствором тиосульфата натрия.
ЦЕРИМЕТРИЯ основана на применении титрованного раствора солей церия (IV), которые в кислой среде восстанавливаются до церия (III)
Се4+ + е = Се3+
Соединения церия (IV) обладают устойчивостью титрованных растворов как при комнатной температуре, так и при нагревании до 100°С и выше.
Метод предложен для определения содержания неорганических соединений, таких, как железо (II), мышьяк (III), и органических лекарственных веществ (углеводов, органических кислот, викасола, производных фенотиазина).
НИТРИТОМЕТРИЯ использует в качестве титранта раствор нитрита натрия. Метод применяется главным образом для определения органических лекарственных веществ. Наибольшее количество методик основано на легко протекающих реакциях диазотирования или нитрозирования. Первичные ароматические амины образуют с нитритом натрия в среде соляной кислоты диазосоединение
Ar—NH2 + NaNO2 + 2HCl → [Ar—N+≡N)C1- + NaCl + 2H20.
Вторичные ароматические амины в таких же условиях образуют нитрозоамины
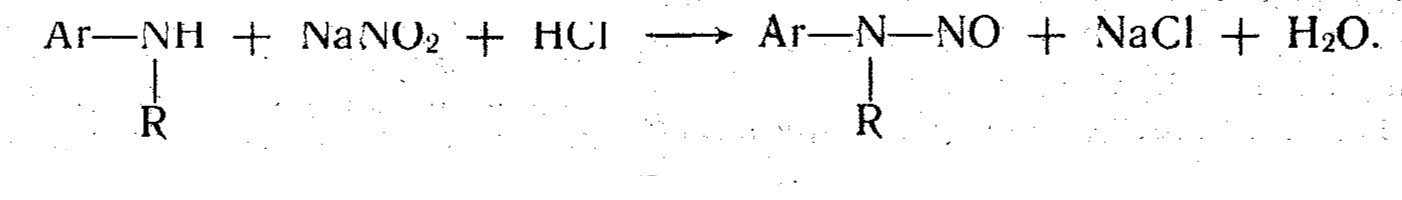
Эквивалентную точку устанавливают с помощью внешних и внутренних индикаторов, а также потенциометрическим методом. Реакция диазотирования является экзотермической и к концу, титрования протекает медленно. Для ускорения в анализируемый раствор прибавляют катализатор — кристаллический бромид калия.
Область применения нитритометрии—определение сульфаниламидных препаратов, производных п-аминобензоиной кислоты.
Расчеты результатов анализа. Содержание вещества (в граммах) в 100 лекарственной формы вычисляют по формуле

где V— объем титрованного раствора; КП — поправочный коэффициент; Т — титр раствора по определяемому веществу, г; а — масса или объем вещества, взятого для анализа.
Содержание вещества (в граммах) во всей массе или во всем объеме лекарственной формы определяют по формуле

где Р — масса или объем лекарственной формы.
Титр раствора по определяемому веществу показывает массу определяемого вещества, которое реагирует с миллилитром титрованного раствора, рассчитывается по формуле
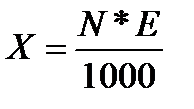
где N—нормальность титрованного раствора; E— величина одного моля эквивалента определяемого вещества.
6.4. Иллюстративный материал слайды с основными пунктами аналитического нормативного документа (АНД) и временного аналитического нормативного документа (ВАНД).
Литература
Основная литература:
1. Арзамасцев А.П. Фармацевтическая химия: учебное пособие, 3-е изд., испр. – М.: ГЭОТАР-Медиа, 2008. – 640 с.
2. Беликов В.Г. Фармацевтическая химия: учебное пособие, 2-е изд. – М.: МЕДпресс-информ, 2008. – 616 с.
3. Руководство к лабораторным занятиям по фармацевтической химии: Э.Н. Аксенова, О.П. Андрианова, А.П. Арзамасцев и др. – М.: Медицина, 2001. – 384 с.
4. Государственная фармакопея Республики Казахстан: первое издание. – 1 том. – Астана: Изд. дом «Жибек жолы», 2008. – 592 с.
Дополнительная литература:
1. Анализ лекарственных смесей / Под ред. А.П. Арзамасцева, В.М. Печенникова, Г.М. Родионова и др. – М.: Компания Спутник+, 2000. – 275 с.
2. Арыстанова Т.А., Ордабаева С.К. Стандартизация лекарственных средств: учебное пособие. – Алматы, 2002. – 98 с.
3. Государственный реестр лекарственных средств. – М.: 2001. – 1277 с.
4. Бейсенбеков А.С., Шаншаров Г.Б., Галымов Е.Г., Бейсенбеков Н.А. Стандартизация лекарств: учебное пособие. – Алматы, 2008. – 167 с.
5. Государственная фармакопея СССР: Х издание. – М.: Медицина, 1968. – 1079 с.
6. Государственная фармакопея СССР: XI издание. – М.: Медицина, 1987. – Т.1. – 334 с.
7. От субстанции к лекарству: учебное пособие / Под ред. чл.-корр. НАН Украины В.П. Черных. – Харьков: изд-во НФаУ «Золотые страницы», 2005. – 1244 с.
Контрольные вопросы
7. Какие критерии оценки качества лекарственных средств Вы знаете?
8. Каким образом можно определить подлинность неорганических лекарственных веществ?
9. Как определяется подлинность органических лекарственных веществ?
10. Как определяют температуру плавления?
11. Поясните методику испытаний на чистоту.
12. Как определяется растворимость лекарственного средства?
Не нашли, что искали? Воспользуйтесь поиском: