ТОР 5 статей:
Методические подходы к анализу финансового состояния предприятия
Проблема периодизации русской литературы ХХ века. Краткая характеристика второй половины ХХ века
Характеристика шлифовальных кругов и ее маркировка
Служебные части речи. Предлог. Союз. Частицы
КАТЕГОРИИ:
- Археология
- Архитектура
- Астрономия
- Аудит
- Биология
- Ботаника
- Бухгалтерский учёт
- Войное дело
- Генетика
- География
- Геология
- Дизайн
- Искусство
- История
- Кино
- Кулинария
- Культура
- Литература
- Математика
- Медицина
- Металлургия
- Мифология
- Музыка
- Психология
- Религия
- Спорт
- Строительство
- Техника
- Транспорт
- Туризм
- Усадьба
- Физика
- Фотография
- Химия
- Экология
- Электричество
- Электроника
- Энергетика
Различные агрегатные состояния растворов. Растворы газов
Когда в повседневной жизни говорят о растворах, обычно имеют в виду жидкие растворы. Однако существуют и растворы газов, и твердые растворы, имеющие кристаллическую структуру. В качестве примера газовых растворов приведем воздух – раствор, состоящий, в основном, из четырех компонентов: азота (около 79 мол. %), кислорода (~20 %), аргона (~1 %) и углекислого газа (0,03 %). Смешение различных газов почти всегда приводит к образованию истинных растворов газов. Однако газы способны смешиваться во всех отношениях не при любых условиях. Возможность неполного смешения газов с образованием двух газообразных фаз, находящихся в равновесии, предвидел еще Ван-дер-Ваальс (1894). Экспериментально ограниченная взаимная растворимость газов была впервые установлена в середине XX века советскими учеными во главе с И.Р.Кричевским в системе аммиак — азот. При высоких давлениях (тысячи атмосфер) гомогенная газообразная система распадается на две газовые фазы с отчетливой границей раздела. К настоящему времени обнаружены и многие другие системы, обладающие этим свойством.
Согласно одной из гипотез, подтвержденной данными некоторых астрономических экспериментов, расслоение газов также имеет место в атмосфере планет-гигантов (Юпитер, Сатурн, Уран, Нептун).
Твердые растворы
При образовании твердых растворов, их состав оказывается варьируем за счет того, что узлы и междоузлия кристаллической решетки могут быть заняты различными компонентами – атомами, молекулами или ионами[1]. Важно отметить, что при образовании таких растворов тип кристаллической решетки растворителя сохраняется. Заметим, что далеко не любой сплав является твердым раствором. Очень часто сплавы металлов оказываются гетерогенными, т.е. по сути – взвесями, распад которых чрезвычайно замедлен по причине очень высоких значений вязкости в твердых телах.
Среди твердых растворов наиболее часто встречаются т.н. твердые растворы замещения, при образовании которых в узлах кристаллической решетки данного вещества атомы, молекулы и ионы замещаются частицами другого компонента. Только для этого типа растворов возможно возникновение непрерывного ряда твердых растворов (т.е. таких, в которых смешение возможно в любой области составов). Непрерывная растворимость встречается не слишком часто и возникает, если исходные компоненты близки по химическим свойствам, им отвечает один и тот же тип кристаллической структуры и число частиц в элементарной ячейке, а размеры частиц компонентов отличаются не более чем на 14—15%. Примеры: в системах Cu‑Au и Ag‑Au при высоких температурах (от 800 °С до плавления) образуется непрерывный ряд твердых растворов замещения, а в системах Cu‑Ag, Sn‑Bi – только ограниченная растворимость по типу замещения.
Помимо твердых растворов замещения существуют также твердые растворы внедрения и вычитания. В двух последних возможна только ограниченная растворимость. Твердые растворы внедрения получаются путем внедрения частиц одного вещества в междоузлия кристаллической решетки другого вещества (растворителя), происходящие без изменения типа этой решетки. Растворы внедрения образуются в том случае, когда размеры частиц внедряемого вещества меньше размеров частиц растворителя. При внедрении новых частиц в промежутки между атомами металла происходит увеличение напряжений в кристаллической решетке. По этой причине область составов, в которой образуются твердые растворы внедрения, обычно невелика. Примеры: Такие объекты обычно возникают при растворении атомов легких неметаллов (водорода, азота, углерода, кислорода, бора, кремния) в металлах (прежде всего – в d- металлах IV, V и VI побочных подгрупп ПС).
Твердые растворы вычитания получаются, когда в структуре сложных твердых веществ[2] в некоторых узлах кристаллической решетки вместо атомов имеются незанятые места – вакансии.
Проще всего заметную концентрацию вакансий в твердом веществе можно создать путем облучения тела частицами с высокими энергиями ( g-, b- лучи и т.п.). В этом случае некоторые атомы просто “выбиваются” со своих позиций. Вместе с тем, некоторая концентрация вакансий (а также межузельных атомов) возникает при любой отличной от абсолютного нуля температуре. При этом, чем выше температура, тем большая равновесная концентрация точечных дефектов (т.е., вакансий и межузельных атомов) присутствует в твердом теле. Таким образом, выращенный даже в самых подходящих для формирования идеальной структуры кристалл всегда является дефектным. Следует заметить, что самопроизвольное достижение равновесной концентрации точечных дефектов осуществляется достаточно медленно даже при высоких температурах (часы и дни при T ~ 103 K ). При быстром охлаждении твердого тела до комнатной или более низких температур почти всегда происходит сохранение достигнутой концентрации дефектов, которая продолжает существовать уже как неравновесная.
Концентрация вакансий, в зависимости от условий синтеза, может изменяться в некоторых пределах. Кроме того, эта концентрация, в общем случае, различается в разных подрешетках. В результате, состав растворов вычитания также может изменяться. В связи с этим такие объекты иногда называют твердыми растворами с дефектной решеткой. Примеры: кислородные, сернистые и мышьяковистые соединения d - металлов, находящихся в низких степенях окисления (Ti1‑aO, Cr1‑bS, Ni1‑gAs).
На примерах твердых растворов вычитания мы видим, что область гомогенности раствора (т.е. области составов, в пределах которой раствор существует как единственная фаза), не всегда включает в себя чистый компонент системы. Например, область гомогенности фазы Ti1‑aO не включает в себя ни чистого титана (x Ti = 1), ни чистого кислорода (x O = 1). Аналогично могут вести себя и твердые растворы внедрения. Если область гомогенности твердого раствора внедрения не захватывает чистого компонента, то обычно говорят о фазе внедрения, кристаллическая структура которой отличается от структуры, в которой кристаллизуется чистый компонент. При насыщении немолекулярного кристалла атомами, способными давать межузельные дефекты при небольших концентрациях примеси твердый раствор внедрения образуется без изменения исходной кристаллической структуры. Если же концентрация примеси превышает некоторую критическую концентрацию, то кристаллическая решетка перестраивается и образуется т.н. фаза внедрения, которая отделена от исходного твердого раствора гетерогенной областью.
Область гомогенности твердых растворов замещения также может быть отделена от каждого из компонентов системы гетерогенными областями. При рассмотрении таких растворов обычно выделяют еще один тип нарушений кристаллической структуры. Антиструктурные дефекты представляют собой атомы, находящиеся в узлах другой подрешетки. Например, в кристалле Ni1-aAl часть атомов алюминия может занимать места, предназначенные кристаллической решеткой для атомов никеля.
Отметим, что в сложных веществах все три типа дефектов всегда присутствуют в твердом теле (пусть некоторые их них – в очень небольших концентрациях). По этой причине твердый раствор относят к одному из трех вышеперечисленных типов по признаку преобладания тех или иных нарушений структуры.
Вакансии, межузельные атомы и антиструктурные дефекты являются т.н. точечными дефектами. Подчеркнем, что только такие дефекты могут быть равновесными. Особенности точечных дефектов состоят в том, что
- во-первых, их концентрацией легко управлять, изменяя условия, в которых находится вещество и,
- во-вторых, даже ничтожная концентрация точечных дефектов способна очень сильно изменять свойства твердых веществ. Таким образом, свойства одного и того же вещества (материала) могут принципиально различаться за счет разной концентрации дефектов.
Жидкие растворы
На практике наиболее часто приходится встречаться с жидкими растворами. При образовании жидкого раствора растворенное вещество может находиться в виде отдельных молекул или ассоциатов, состоящих из нескольких молекул, либо частично или полностью подвергаться ионизации (диссоциации) на ионы. В связи с этим жидкие растворы подразделяются на растворы неэлектролитов и растворы электролитов.[3]
Электролиты и неэлектролиты
Согласно представлениям о химических взаимодействиях, все растворы могут быть разделены на две большие группы – растворы электролитов и неэлектролитов. В электролитах растворенная часть вещества подвергается – при непосредственном участии растворителя – полной или частичной ионизации (диссоциации) с образованием сольватированных ионов (т.е. ионов, окруженных молекулами или атомами растворителя). Например:
 , (I.4)
, (I.4)
где символ “ aq ” подчеркивает сольватированное (в данном случае – гидратированное) состояние ионов.
Различают слабые и сильные электролиты. Для последних ионизационное равновесие очень сильно смещено вправо, т.е. растворенное вещество практически полностью превращается в ионы. Из водных сред к сильным электролитам относятся растворы почти всех солей (за исключением солей кадмия, ртути и некоторых других тяжелых металлов), многие кислоты (например, хлорная, азотная, галогеноводородные – HCl, HBr и HI, трифторноуксусная CF3COOH) и основания (едкие щелочи: NaOH, KOH, азотистые основания ([N(CH3)4]OH) и др.).
К числу наиболее убедительных доказательств практически полной ионизации сильных электролитов относятся следующие.
1. При пропускании фотонов через поглощающий свет раствор (не обязательно в видимом глазом диапазоне) интенсивность поглощения света пропорциональна концентрации предполагаемых ионов. Если бы реакция ионизации была бы заметно обратимой, то интенсивность поглощения более слабо зависела от концентрации, поскольку разбавление увеличивает степень ионизации.
2. При нейтрализации водных растворов сильных кислот (HNO3, HClO4, HCl, HBr, HI и др.) сильными основаниями (NaOH, KOH и др.) независимо от природы кислоты и основания выделяется одно и то же количество теплоты (~56 кДж/моль). Это можно объяснить протеканием в растворе одной и той же реакции:
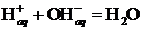 , (I.5)
, (I.5)
где символ “ aq ” означает гидратированное состояние ионов.
3. Реакции сильных электролитов с образованием малодиссоциированных веществ (например, I.5.) протекают с очень высокой скоростью, которая (при очень интенсивном перемешивании растворов) почти не зависит от температуры. Расчеты показывают, что это возможно лишь в случае, когда практически любое соударение взаимодействующих частиц приводит к образованию молекул продукта, а это, в свою очередь, возможно только при полной ионизации исходных веществ.
В противоположность перечисленным, существует группа растворов, для которых реакция с образованием ионов является заметно обратимой. Это электролиты средней силы, либо слабые. К первым принадлежат, например, растворы плавиковой (HF), сернистой (H2SO3), муравьиной (HCOOH) кислот, гидроксидов лития и магния. К слабым электролитам относят некоторые кислоты (для водных растворов это H2CO3, H3BO3, C6H5OH и др.) и основания (Al(OH)3, NH3, C6H5NH2 и др.), а также некоторые комплексные соединения ([Fe(H2O)3(CNS)3] и др.). В случае электролитов средней силы и слабых электролитическое равновесие удобно характеризовать константой ионизации, которая является частным случаем константы равновесия (KC) и для разбавленных растворов связывает концентрации (Сi) различных ионных и молекулярных форм в растворе согласно закону Гульберта-Вааге. Важно подчеркнуть, что сама константа от этих концентраций не зависит (для разбавленных растворов). Например, для разбавленного водного раствора синильной кислоты (HCN) имеем:
 , (I.6)
, (I.6)
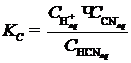 (I.7)
(I.7)
При стандартных условиях величина KC (HCN) = 7,9×10‑9.
Обычно говорят об электролите средней силы, если величина константы ионизации (диссоциации) находится в пределах от 10‑4 до 102. Слабые электролиты характеризуются величиной KC, меньшей 10‑4.
Ряд веществ при растворении подвергается ступенчатой ионизации. Например, для водных растворов орто-фосфорной кислоты при стандартных условиях имеем:
 , (I.8)
, (I.8)
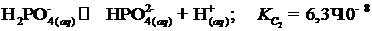 , (I.9)
, (I.9)
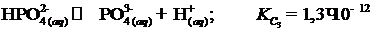 . (I.10)
. (I.10)
Таким образом, орто-фосфорная кислота является кислотой средней силы, если говорить о первой ступени ионизации и слабой кислотой, если рассматривать две последующие ступени ионизации.
Общепринятым считается относить водный раствор серной кислоты к сильным электролитам. Однако это так, если речь идет об ионизации по первой ступени. При рассмотрении ионизации водной H2SO4 по второй ступени, этот электролит будет относиться к кислотам средней силы, т.к. KC 2 = 1,2×10‑2.
Для сильных электролитов также может быть формально введена величина KC и записано выражение для ее связи с концентрациями молекул и ионов. Однако практически использовать такое выражение не представляется возможным (подумайте, почему).
Сила электролитов и способ их ионизации очень сильно зависят от природы растворителя. Например, азотная кислота, растворенная в серной кислоте, ведет себя как слабое основание, т.к. связывает ионы H+, возникающие при ионизации серной кислоты. Еще пример. Бензол, немного растворимый в воде, практически не подвергается ионизации в таких растворах. Однако раствор бензола в безводном фтороводороде уже является слабым электролитом.
Наконец, существует группа растворов, которые практически не содержат ионов. Их называют неэлектролитами. Заметим, что излагаемые ниже элементарные представления о теориях растворов были изначально сформулированы для неэлектролитов.
II. О ТЕОРИЯХ РАСТВОРОВ
Физическая теория растворов, возникшая в конце XIX в., явилась обобщением ряда исследований, главным образом, Я. Вант-Гоффа, С. Аррениуса и В. Оствальда. Эта теория опиралась на экспериментальное изучение свойств, в основном, разбавленных растворов, а также растворов веществ очень близких как по химической природе, так и по физическим свойствам (например, раствор в системе н-гептан – метилциклогексан). Было выявлено, что многие свойства таких растворов, зависят от концентрации растворенного вещества, а не от его природы. Количественные законы (законы Я.Вант-Гоффа, Ф.Рауля) были интерпретированы в предположении, что в идеальных растворах поведение молекул (атомов) растворенного вещества подобно поведению таких частиц в идеальном газе. Отступления от этих законов, наблюдаемые для растворов электролитов, были объяснены на основе теории электролитической диссоциации С.Аррениуса. Аналогия между сильно разбавленными растворами и газами многим ученым показалась столь убедительной, что они стали рассматривать растворение как исключительно физическое явление, в котором растворитель является только средой, в которую могут диффундировать частицы растворенного вещества. Простота представлений физической теории растворов и успешное применение ее для объяснения многих свойств растворов обеспечили быстрый успех этой теории. Однако достаточно концентрированные растворы демонстрировали явное несоответствие между теорией и экспериментом. По этой причине возникла необходимость дальнейшего развития представлений о растворах.
Химическая теория растворов, созданная преимущественно Д.И. Менделеевым и его последователями, рассматривала процесс образования раствора как разновидность химического процесса, для которого характерно взаимодействие между частицами компонентов. Д.И. Менделеев рассматривал растворы как системы, образованные частицами растворителя, растворенного вещества и неустойчивых химических соединений, которые образуются между ними и находятся в состоянии частичного и обратимого распада, а реакции в растворах, протекают в соответствии с законом действующих масс. Он также указывал на необходимость использования всей суммы физических и химических сведений о свойствах частиц, образующих раствор, подчеркивал, что все компоненты раствора равноправны и без учета свойств и состояний каждого из них нельзя дать полной характеристики системы в целом. Большое значение ученый придавал изучению свойств растворов как функции температуры, давления, концентрации; он впервые высказал мысль о необходимости изучения свойств растворов в смешанных растворителях. Развивая учение Д.И.Менделеева, сторонники химического взгляда на природу растворов, указывали, что частицы растворенного вещества движутся не в пустоте, а в пространстве, занятом частицами растворителя, с которыми они взаимодействуют, образуя сложные, различные по устойчивости соединения.
Развитие химической теории растворов шло в нескольких направлениях, объединенных единой идеей о взаимодействии растворителя с растворенным веществом. Эти исследования касались нахождения определенных соединений в растворе на основе изучения диаграмм состав — свойство, изучения давления пара над растворами, распределения веществ между двумя растворителями, изучения термохимии растворов. Работы по определению соединений в растворах были связаны с большими трудностями, так как прямым опытом было невозможно доказать существование ассоциатов (гидратов) в водных растворах, поскольку они находятся в состоянии химического равновесия с продуктами своего распада, и попытки выделить их из растворов в свободном виде заканчивались неудачей. Большое значение для подтверждения химической теории растворов имели термохимические исследования. На многих системах было показано, что при образовании раствора наблюдается охлаждение или нагревание системы, что объясняли химическим взаимодействием между компонентами. Химическую природу процесса растворения подтверждали и исследования давления пара над раствором, и изучение распределения веществ между двумя растворителями.
Следует заметить, что если к концентрированным растворам неэлектролитов иногда могут быть применены соотношения из физической теории растворов в максимально упрощенном виде (соотношения для идеальных растворов), то этого почти никогда нельзя сделать для заметно диссоциирующих электролитов.
К началу XX в. накопился обширный экспериментальный материал, показывающий, что растворы являются сложными системами, в которых наблюдаются явления ассоциации, диссоциации, комплексообразования, и при их изучении необходимо учитывать все виды взаимодействия между частицами, имеющимися и образующимися в растворе. В связи с большим разнообразием растворов для объяснения их природы и свойств часто используются представления как физической, так и химической теории растворов.
Термодинамические условия образования растворов
Для простоты рассмотрим образование раствора в бинарной (т.е. двухкомпонентной) системе из двух исходно однокомпонентных фаз. Образование раствора из компонентов — процесс самопроизвольный, в котором, как и в любом самопроизвольном процессе, протекающем в закрытой системе, находящейся под влиянием двух внешних факторов (Р и T), D G < 0. Следовательно, термодинамическим условием образования раствора является убыль энергии Гиббса. Такой процесс будет протекать самопроизвольно до тех пор, пока в системе иссякнет запас одной из фаз (образование ненасыщенного раствора), либо – пока растворы не станут насыщенными и не установятся равновесия:
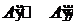 , (II.1)
, (II.1)
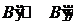 . (II.2)
. (II.2)
где A и B – компоненты, находящиеся изначально в фазах (¢) и (²).
При равновесии D G = 0. Исключение составляют термодинамически неустойчивые пересыщенные растворы. С точки зрения термодинамики раствор называется насыщенным, когда химический потенциал чистого растворяемого вещества (твердого, жидкого или газообразного) равен химическому потенциалу этого вещества в растворе.
Рассмотрим раствор, состоящий из растворителя А и растворенного вещества В. Пусть энергия Гиббса чистого компонента А количеством 1 моль равна 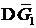 , чистого компонента В (количеством также 1 моль) равна
, чистого компонента В (количеством также 1 моль) равна 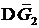 . Если раствор образуется из n 1 моль А и n 2 моль В, то общая энергия Гиббса системы, состоящей из двух компонентов, до смешения будет равна:
. Если раствор образуется из n 1 моль А и n 2 моль В, то общая энергия Гиббса системы, состоящей из двух компонентов, до смешения будет равна:
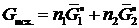 , (II.3)
, (II.3)
так как энергия Гиббса — экстенсивное свойство.
Отнесем энергию Гиббса системы до смешения компонентов к одному молю:
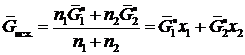 , (II.4)
, (II.4)
где x 1 и x 2 — молярные доли.
Следовательно, молярная энергия Гиббса системы, состоящей из двух компонентов, до смешения линейно зависит от состава этой системы. Таким же значением энергии Гиббса будет характеризоваться механическая смесь различных фаз в том случае, если вещества абсолютно не растворяются друг в друге.
Если компоненты А и В образовали гомогенный раствор, то последний будет иметь иное значение энергии Гиббса. Энергия Гиббса получившегося при смешении раствора, отнесенная к 1 молю (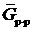 ) также зависит от состава системы, но изменяется нелинейно. (Под молем раствора понимается такое количество раствора, в котором число молей каждого компонента равно его молярной доле.) Изменение энергии Гиббса при образовании моля бинарного раствора равно:
) также зависит от состава системы, но изменяется нелинейно. (Под молем раствора понимается такое количество раствора, в котором число молей каждого компонента равно его молярной доле.) Изменение энергии Гиббса при образовании моля бинарного раствора равно:
 . (II.5)
. (II.5)
С изменением состава раствора 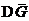 изменяется, но во всех случаях
изменяется, но во всех случаях  .
.
Уменьшение энергии Гиббса в процессе образования раствора может быть обусловлено энтропийным, либо совместным энтальпийным и энтропийным вкладом (значителен “химический” вклад), так как
 (II.6)
(II.6)
Особенность термодинамического подхода к растворам заключается в том, что здесь не требуется представлений о молекулярном механизме взаимодействия в растворах. Основным вопросом при таком подходе является установление зависимости свойств, растворов от состава и свойств его компонентов. Для установления такой зависимости пользуются парциальными молярными величинами, прежде всего, химическим потенциалом m i.
Идеальные растворы
Выше отмечалось, что многие свойства разбавленных растворов, а также растворов веществ с очень близкой химической природой зависят от числа молей растворенного вещества (т.е., от присутствующего числа частиц примеси). Напомним, что именно на таких растворах была развита физическая теория растворов, в которой центральное место занимает аналогия между растворами (любого агрегатного состояния) и идеальными газами. Было постулировано, что концентрационная зависимость химического потенциала компонента раствора должна быть такой же, как и для смеси идеальных газов:
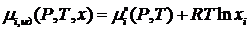 (II.7)
(II.7)
(Выражение II.7 получило название “гипотезы Льюиса” или “представления Льюиса”.)
Рассмотрим, какими важнейшими макроскопическими признаками характеризуется образование идеального раствора. Напомним, что
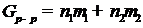 и
и 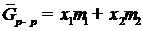 (II.8)
(II.8)
Тогда с учетом (II.5) и (II.7) изменение энергии Гиббса при образовании 1 моля идеального раствора записывается как
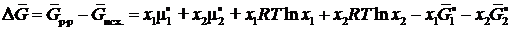 (II.9)
(II.9)
Изменение энергии Гиббса в расчете на моль смеси данного состава называют энергией (Гиббса) смешения (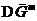 ). С учетом того, что
). С учетом того, что 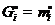 (II.9) преобразуется в (II.10):
(II.9) преобразуется в (II.10):
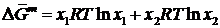 , (II.10)
, (II.10)
Отметим по ходу, что, так как  , то <0, т.е. самопроизвольный процесс образования идеального раствора сопровождается убылью энергии Гиббса.
, то <0, т.е. самопроизвольный процесс образования идеального раствора сопровождается убылью энергии Гиббса.
Чтобы определить изменение энтальпии при образовании идеального раствора, заметим, что в правой части находятся величины, не зависящие от температуры, поэтому
 (II.11)
(II.11)
В классической термодинамике существует уравнение Гиббса-Гельмгольца, одна из форм записи которого выглядит как
 . (II.12)
. (II.12)
С учетом того, что T ¹0, получаем
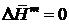 , (II.13)
, (II.13)
т.е., энтальпия образования идеального раствора из компонентов равна нулю.
Далее отметим, что правая часть уравнения (II.9) не зависит от давления, т.е.
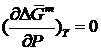 (II.14)
(II.14)
Но частная производная энергии Гиббса по давлению представляет собой объем. Тогда получается, что при образовании идеальных растворов отсутствуют не только тепловые, но и объемные эффекты (расширения/сжатие):
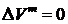 . (II.15)
. (II.15)
Однако изменение энтропии при образовании идеальных растворов оказывается отличным от нуля. Поскольку
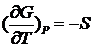 , (II.16)
, (II.16)
то с учетом (II.9) получаем
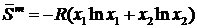 . (II.17)
. (II.17)
Так как мольные доли компонентов находятся в промежутке от нуля до единицы, то энтропия смешения для идеальных растворов оказывается положительной (иначе не могло и быть, поскольку тепловой эффект равен нулю, а общее изменение энергии Гиббса должно быть меньше нуля).
Не нашли, что искали? Воспользуйтесь поиском: