ТОР 5 статей:
Методические подходы к анализу финансового состояния предприятия
Проблема периодизации русской литературы ХХ века. Краткая характеристика второй половины ХХ века
Характеристика шлифовальных кругов и ее маркировка
Служебные части речи. Предлог. Союз. Частицы
КАТЕГОРИИ:
- Археология
- Архитектура
- Астрономия
- Аудит
- Биология
- Ботаника
- Бухгалтерский учёт
- Войное дело
- Генетика
- География
- Геология
- Дизайн
- Искусство
- История
- Кино
- Кулинария
- Культура
- Литература
- Математика
- Медицина
- Металлургия
- Мифология
- Музыка
- Психология
- Религия
- Спорт
- Строительство
- Техника
- Транспорт
- Туризм
- Усадьба
- Физика
- Фотография
- Химия
- Экология
- Электричество
- Электроника
- Энергетика
Работа 4. Определение свежести мяса и рыбы
Мясную ткань измельчают, берут 2 г фарша в пробирку, смешивают с 2 мл дистиллированной воды, встряхивают, дают отстояться и отделяют верхний водяной слой. В УФ-свете наблюдают свечение экстракта. У свежего мяса – свечение экстракта желто-зеленое, сомнительной свежести –зелено-голубое, несвежего- светло-голубое, молочное. У экстракта свежей рыбы свечение фиолетовое, сомнительной свежести—фиолетово-синее, У несвежей рыбы- синее.
ПОЛЯРИМЕТРИЯ
Поляриметрический метод основан на свойстве некоторых веществ изменять направление поляризованных лучей света.
Вещества, обладающие свойством изменять направление колебаний при прохождении через них поляризованного света, называются оптически анизотропными, или оптически активными, в отличие от оптически изотропных, или неактивных, которые этих изменений не вызывают.
Оптическая активность веществ обусловлена особенностями строения кристаллической решетки – в этом случае вещества проявляют оптическую активность только в твердом кристаллическом состоянии, или особенностями строения молекул – оптическая активность таких веществ проявляется только в растворах.
К веществам последней группы относятся главным образом такие органические вещества, как сахароза, фруктоза, глюкоза, винная кислота. Поляриметрический метод разработан для количественного определения веществ именно этой группы.
У поляризованного луча, пропущенного через слой раствора оптически активного вещества, плоскость поляризации оказывается повернутой на некоторый угол, называемый углом поворота плоскости поляризации.
Плоскость, проходящая через поляризованный луч перпендикулярно направлению его колебаний, называется плоскостью поляризации.Угол поворота плоскости поляризации зависит от природы вещества, концентрации его в растворе, толщины слоя раствора, через который проходит поляризованный луч, а также длины волны поляризованного луча и температуры.
Оптическая активность вещества характеризуется удельным вращением, под которым понимают угол, на который повернется плоскость поляризации при прохождении поляризованного луча через раствор, в 1 мл которого содержится 1 г растворенного вещества, при толщине слоя раствора (длине поляризационной трубки), равной 1 дм.
Удельное вращение зависит не только от природы вещества, но и от температуры, длины волны поляризованного света и растворителя, поэтому его принято относить к температуре 20 °С и желтой линии натрия и обозначать  с указанием растворителя.
с указанием растворителя.
Угол вращения плоскости поляризации [α] определяют по формуле
 , (1.9)
, (1.9)
где 1 – длина трубки, дм; с – концентрация вещества, г/100 мл; σ – удельное вращение, град.
Пользуясь формулой (1.9), легко вычислить количество вещества в граммах, содержащееся в 100 мл раствора, т. е. концентрацию (с).
 .
.
Основные рабочие части поляриметра: устройство для поляризации света – поляризатор; устройство для определения угла поворота плоскости поляризации после прохождения поляризованных лучей через исследуемый раствор – анализатор; поляризационная трубка, наполняемая исследуемым раствором и помещаемая между поляризатором и анализатором.
В качестве поляризаторов применяют кристаллы исландского шпата или других минералов, обладающих свойством двойного лучепреломления.
Анализатор в отличие от поляризатора может поворачиваться вокруг оптической оси прибора. Когда главные линии призм анализатора и поляризатора расположены параллельно, свет, проходящий через поляризатор, пройдет и через анализатор (при отсутствии между ними оптически активного раствора).
Если же главные сечения анализатора и поляризатора взаимно перпендикулярны, лучи через анализатор не пройдут.
При всех промежуточных положениях через анализатор от поляризатора пройдет только часть лучей.
Если поместить между поляризатором и анализатором оптически активное вещество, то часть поляризованных лучей при прохождении через оптически активный раствор отклонится и в анализатор не попадет. Поэтому световое поле получит неоднородную окраску.
Вращая анализатор, добиваются исходного (до помещения раствора) положения анализатора и по углу поворота прибора, соединенного со шкалой, судят о степени вращения плоскости поляризации исследуемым раствором, что позволяет рассчитать количество вещества в растворе.
Различают поляриметры с установкой на полную темноту и полутеневые с двойным и тройным полем зрения. Наиболее широкое применение получили полутеневые поляриметры. У поляриметров этого типа с двойным полем зрения поляризатор состоит из двух николей, а у поляриметров с тройным полем зрения – из трех. Разновидностью поляриметра является сахариметр, предназначенный для определения содержания сахарозы в растворах. В отличие от поляриметров других видов сахариметр имеет линейную шкалу, градуированную по сахарозе. 100° этой шкалы соответствуют 34, 62 кругового радиуса шкалы поляриметра.
Сахариметр показывает 100°, если в трубке длиной 200 мм поляризуют раствор, в 100 мл которого при 20 °С содержится точно 26 г химически чистой, абсолютно сухой сахарозы.
Особенность оптической системы сахариметра состоит в том, что анализатор в нем поставлен на полутень по отношению к поляризатору и укреплен неподвижно. Изменение угла поворота плоскости поляризации, вызванное исследуемым раствором, устанавливают клиновым кварцевым компенсатором.
Кварц – оптически активное вещество, обладающее вращательной способностью, близкой к вращательной способности сахарозы, но обратной по направлению. Меняя толщину слоя кварца, можно полностью компенсировать вращение, вызываемое сахарозой.
Существует несколько типов компенсаторов, отличающихся один от другого количеством кварцевых клиньев и пластинок. Но принцип действия их одинаков и основан на том, что относительным перемещением клиньев можно менять толщину кварцевого слоя.
Подвижный клин (или клинья) смешается шестеренкой, соединенной с подвижной горизонтальной шкалой, что позволяет отсчитывать непосредственно концентрацию раствора сахарозы.
Приготовление раствора для сахариметра. На аналитических весах отвешивают точно 26 г сахара-песка или предварительно измельченного в ступке сахара-рафинада. Навеску переносят в химический стакан, куда небольшими порциями добавляют теплую дистиллированную воду. Полученный раствор с помощью воронки и стеклянной палочки без потерь переливают в мерную, колбу вместимостью 100 мл. Стакан и воронку тщательно ополаскивают и промывные воды также сливают в мерную колбу. Раствор доливают до метки только после того, как температура его и дистиллированной воды достигнет 20 °С. После тщательного взбалтывания содержимого колбы раствор фильтруют через бумажный фильтр. В процессе фильтрования во избежание испарения воды и нарушения концентрации раствора воронку необходимо покрыть часовым стеклом. Сразу по окончании фильтрования раствор следует подвергнуть поляриметрии.
Порядок проведения анализа. Трубку сахариметра (рис. 1), только что бывшую в употреблении (не сухую), сначала ополаскивают дистиллированной водой, а затем двумя-тремя небольшими порциями испытуемого раствора. Торцевые (покрывные) стекла промывают дистиллированной водой и досуха протирают.
При исследовании чистого сахара-песка или рафинада пользуются трубкой длиной 200 мм, для сметок – 100 мм.
Трубку, закрытую с одной стороны, наполняют так, чтобы верхний мениск раствора выступал над ее краями, после чего трубку накрывают стеклышком, а затем уже навинчивают металлическую головку. Далее проверяют, нет ли в трубке пузырьков воздуха. При наличии их головку со стеклом снимают и в трубку добавляют несколько капель раствора. С наружной стороны торцовые стекла должны быть абсолютно сухими, иначе поле зрения окажется затемненным.

Рис. 1. Сахариметр типа СУ-3:
1 – лупа в оправе; 2 – узел измерительной головки; 3 – винт установки шкалы на нуль; 4 – съемный ключ; 5 – камера для поляриметрических кювет;6 – поляризатор; 7 – поворотная обойма со светофильтром; 8 – осветительный узел; 9 – винты для закрепления патрона с лампой; 10 – траверса; 11 – вилка разъема; 12 – винт заземления; 13 – основание; 14 – рукоятка кремальерной передачи: 15 – гильза с анализатором; 16 – зрительная труба.
Перед тем, как поместить трубку в камеру, окуляр зрительной трубы и лупу шкалы вращением их оправ устанавливают на максимальную резкость изображения так, чтобы были четко видны вертикальная линия, разделяющая поле зрения на две половины, а в поле зрения лупы – штрихи и цифры шкалы и нониуса.
Перед началом измерений прибор устанавливают на нуль. Для этого (при отсутствии в камере поляриметрической кюветы) вращением рукоятки кремальерной передачи добиваются полной однородности обеих половин поля зрения, при этом нулевые деления шкалы и нониуса должны совпасть. В противном случае посредством ключа перемещают нониус до совмещения его нулевого деления с нулевым делением шкалы, после чего приступают к измерениям.
В камеру прибора вкладывают поляриметрическую кювету с испытуемым раствором, в результате изменяется однородность обеих половин поля зрения. Вращением рукоятки кремальерной передачи уравнивают их освещенность. После этого, пользуясь нониусом, отсчитывают показания с точностью до 0,5°. Затем проверяют уравнивание освещенностей обеих половин поля зрения и снова отсчитывают показания (рис. 2).

Рис. 2. Положение шкалы и нониуса, соответствующее отсчету:
а – 11,8°; б – 3,2°.
Отсчет производят не менее трех раз; при этом каждый отсчет начинают с возвращения рукоятки в нулевое положение. Записав все три показания (P1, Р2, Р3), рассчитывают среднее из них (Рср) по формуле

где Рср – содержание сахарозы в исследуемом продукте, % (при использовании трубки длиной 100 мм результат умножают на 2).
Содержание сахарозы в сахаре принято выражать в пересчете на сухое вещество, поэтому определяют также влажность сахара путем его высушивания.
Зная влажность сахара (w), содержание сахарозы (X) в процентах в пересчете на сухое вещество вычисляют по формуле

Сахариметром можно определить содержание сахарозы в растворе неизвестной концентрации без предварительного взятия навески. При этом сначала определяют плотность исследуемого вещества (раствора), а затем вычисляют содержание сахарозы в процентах по формуле

РЕФРАКТОМЕТРИЯ
Цель работы: освоить методы определения сухих веществ и коэффициента преломления различных веществ и продовольственных товаров методом рефрактометрии.
Рефракция- это явление преломления луча света на границе раздела двух сред, различных по оптической плотности. Явления лучепреломления или рефракция луча света. наступает тогда, когда луч света, направленный наклонно к плоскости раздела двух сред, переходит из одной среды в другую(скорость распространения света в этих средах не одинакова).
Рефрактометрию широко применяют при исследовании жиров, томатных продуктов, варенья, джема. Этим методом пользуются также для количественного определения жиров в пищевых продуктах, влажности, содержания спирта в растворе (в сочетании с пикнометрическим методом), для по фазного контроля в процессе производства пищевых продуктов – кондитерских, напитков, некоторых видов консервов и т. д.
Показатель преломления зависит от температуры, поэтому рефрактометрические измерения принято выполнять при 20 °С. При отклонении температуры от 20 °С вводят соответствующие температурные поправки.
При работе с раствором следует учитывать, что между показателем преломления и процентным содержанием вещества в растворе не всегда существует прямая зависимость. Поэтому судить о концентрации вещества в растворе по показателю преломления можно только при наличии кривых, выражающих зависимость между этими двумя величинами. В некоторых случаях по показателю преломления невозможно определить содержание вещества в растворе, так как даже при значительных колебаниях концентрации вещества показатель преломления изменяется очень мало (например, для растворов метилового спирта). При наличии в растворе двух веществ только по показателю преломления нельзя судить о состоянии системы. В этом случае требуется знать какие-либо другие физико-химические величины, например температуру кипения или плавления, плотность вещества.
Для измерения показателя преломления жидких веществ и растворов применяют приборы, называемые рефрактометрами. Большинство рефрактометров устроено так, что исследуемое вещество помещается между двумя призмами (двумя половинами призмы). Свет, пропущенный через призму, преломляясь или отражаясь от границы раздела сред (призма – вещество), освещает только часть шкалы, образуя достаточно резкую границу света и тени. Положение этой границы на шкале зависит от угла полного внутреннего отражения исследуемого вещества. На шкале указаны показатели преломления, соответствующие различным значениям угла полного внутреннего отражения.
В качестве источника света пользуются монохроматическим светом натриевой горелки или обычным дневным светом, направляемым специальным зеркалом. Поскольку дневной свет состоит из лучей различной длины волн, при прохождении его через призму возникает явление светорассеяния (дисперсии), в результате чего на границе света и тени образуется радужная полоска, затрудняющая отсчет по шкале рефрактометра. Поэтому во всех современных конструкциях рефрактометров предусматриваются компенсаторы, позволяющие устранить дисперсию света.
Компенсаторы – это оптические системы, представляющие собой совокупность двух или трех призм или линз, изготовленных из стекла разных видов и установленных так, что различные цвета спектра налагаются один на другой, благодаря чему граница света и тени становится отчетливой. Поскольку световые лучи исходят из всех точек поверхности призмы и в различных направлениях, на пути между призмой и шкалой помещают линзу, собирающую все параллельные лучи в фокус. Таким образом, каждому пучку световых лучей, прошедших через линзу, соответствует световая точка.
Температуру призмы и исследуемого вещества контролируют термометрами, вмонтированными в оправу призм. Для регулирования и поддержания постоянной температуры в полые металлические оправы призм по специальным устройствам подают воду требуемой температуры. При отсутствии этих устройств пользуются таблицами температурных поправок.Правильность показаний рефрактометров проверяют контрольной жидкостью или дистиллированной водой, показатель преломления которой при 20 °С равен 1,333, либо пользуются котировочной пластинкой, на которой указан показатель преломления. При наложении пластинки на призму рефрактометра показание шкалы должно совпадать с показателем, указанным на пластинке.
Рефрактометр ИРФ-22. Этот прибор (рис. 3) применяют для измерения показателей преломления как жидких, так и твердых продуктов в интервале 1,3-1,7. Основные рабочие части прибора: две призмы, сложенные гипотенузными сторонами и составляющие вместе с оправами полушаровидной формы измерительную головку, зрительная труба с отсчетным устройством.
В оправах призм имеются камеры, через которые пропускают воду и таким образом регулируют температуру призм и заключенного между ними вещества. Температуру контролируют термометром, ввинченным в оправу призм. Для устранения радужной полоски на границе раздела темной и светлой частей поля зрения предназначен компенсатор. Шкала отсчетного устройства жестко соединена с измерительной головкой и помещена в корпусе прибора.
Рефрактометр снабжен двумя зеркалами: одно служит для направления света через исследуемое вещество и призмы, другое – для освещения шкалы показателей преломления.
Принцип действия рефрактометра. Лучи света, отражаясь от зеркала, проходят через призму, гипотенузнаясторона которой матовая, благодаря чему происходит рассеивание света. В исследуемое вещество попадают лучи различных направлений, в том числе и скользящие. Эта призма является осветительной. Пройдя слой жидкости и преломившись на границе жидкость – стекло, лучи войдут во вторую призму (измерительную). Максимальным углом выхода лучей из призмы будет предельный угол, соответствующий скользящим лучам. Таким образом, поле зрительной трубы, поставленной на пути лучей, окажется разделенным на две части – темную и светлую. При этом положение границы раздела будет определяться предельным углом, зависящим от природы вещества.

Рис. 3. Рефрактометр ИРФ-22:
1 – зрительная труба с отсчетным устройством; 2 – корпус; 3 – барабан со шкалой;4 – маховичок для вращения призмы; 5 – шланг; 6 – измерительная головка; 7 – корпус термометра; 8 – штуцеры; 9 – зеркало.
Порядок проведения анализа. С помощью контрольной жидкости или дистиллированной воды проверяют правильность показаний рефрактометра. Затем, протерев досуха призмы, оплавленной стеклянной палочкой на нижнюю призму помещают несколько капель исследуемой жидкости, из которой предварительным фильтрованием через вату или марлю удаляют взвешенные частицы. Нижнюю призму накрывают верхней. Смотря в окуляр, зеркалом направляют отраженный свет в верхнюю часть окошка оправы призмы – если жидкости не окрашены, и в нижнюю часть – если жидкости темноокрашенные и мутные. Затем рычагом перемещают окуляр до совмещения визира с границей раздела темной и светлой частей поля зрения. Головку окуляра необходимо установить по глазу так, чтобы деления и цифры шкалы были отчетливо видны. Если граница окажется расплывчатой, радужной, то, вращая рукоятку компенсатора, добиваются четкости и записывают показания шкалы. Таким образом производят не менее трех отсчетов. Окончательным результатом является среднее арифметическое из всех отсчетов. В процессе определения необходимо следить за показаниями термометра. Если температура окажется выше или ниже 20 °С, то необходимо пользоваться температурными поправками либо произвести предварительное темперирование призм.
Простейшим устройством для темперирования призм является тубусная склянка вместимостью 3-5 л, соединяемая резиновыми трубками с оправами призм и установленная несколько выше рефрактометра. Вода, имеющая температуру на 2-3° выше 20 °С (в зависимости от температуры помещения), сначала поступает в оправы, а затем через трубку, надетую на нижний отросток оправы, в раковину или ведро. Скорость поступления и вытекания воды в оправы регулируют винтовыми зажимами.Для темперирования можно использовать ультратермостаты ТС-15 или другого типа.
После того, как граница раздела света и тени совместится с перекрестием трубы, делают отсчет по шкале. Индексом для отсчета служит неподвижный горизонтальный штрих. Целые, десятые, сотые и тысячные доли показателя преломления отсчитывают по шкале, десятитысячные доли оценивают на глаз.
Рефрактометр РПЛ -3. Этот рефрактометр (рис. 4) предназначен для определения показателя преломления жидкости и содержания сухих веществ по сахарозе в кондитерских изделиях, консервах, крахмале, патоке и т. д.
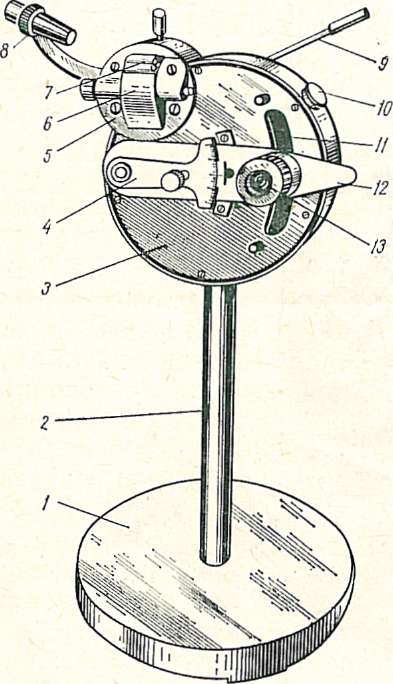
Рис. 4. Рефрактометр РПЛ-3:
1 – основание;2 – колонка; 3 – корпус; 4 – дисперсионный лимб с ручкой; 5 – нижняя камера;6 – шарнир соединения верхней и нижней камер; 7 – верхняя камера; 8 – осветитель; 9 – термометр в оправе; 10 – пробка для ввода ключа для установления нуля; 11 – шкала; 12 – рукоятка; 13 – окуляр.
Перед началом измерений проверяют нуль-пункт прибора, для чего на полированную плоскость измерительной призмы наносят 1-2 капли дистиллированной воды и устанавливают окуляр на резкость по шкале и визирной линии сетки. После этого окуляр перемещают по шкале до тех пор, пока визирная линия сетки не совместится с границей светотени. При правильной установке прибора на нуль-пункт граница светотени при 20 °С должна совместиться с нулевым делением шкалы сухих веществ и делением n0= 1,33299 шкалы показателей преломления. В случае отклонения от этих значений прибор необходимо установить ключом на нуль, для чего следует освободить пробку на корпусе, через отверстие в корпусе на квадрат винта вставить ключ и вращением его в ту или другую сторону совместить линию границы светотени с делением n0= 1,33299, нулевым делением шкалы сухих веществ и визирной линией сетки.
Установив прибор на нуль-пункт, поднимают верхнюю камеру, вытирают соприкасающиеся плоскости призмы досуха сначала фильтровальной бумагой, а затем не ворсистой салфеткой. После этого на поверхность измерительной призмы наносят 1-2 капли исследуемого раствора и плавно опускают верхнюю камеру.
В одно из окон осветителем направляют свет, при этом другое окно должно быть закрыто ширмой. Перемещая окуляр, вводят в поле зрения прибора границу светотени, устанавливают ее на резкость, одновременно поворачивают сектор компенсатора. Перемещают рукоятку с окуляром до совмещения визирной линии сетки с границей светотени и по шкале производят отсчет показаний.
ФОТОКОЛОРИМЕТРИЯ
Задача фотоколориметрии – определение содержания вещества в растворе. Фотоколориметрический метод основан на избирательном поглощении исследуемым веществом монохроматического света. Окраска исследуемого растворенного вещества может быть естественной или полученной при взаимодействии его со специфическими реактивами.
В фотоэлектроколориметрах в отличие от спектрофотометров монохроматический свет выделяют окрашенными светофильтрами в довольно широком участке спектра.
Изменение интенсивности светового потока при прохождении его через окрашенное вещество измеряют фотоэлементами. Каждое окрашенное вещество характеризуется спектром поглощения.
Содержание исследуемого вещества определяют непосредственно в испытуемом растворе или после предварительного отделения его от тех примесей, которые также могут образовывать окрашенные соединения с добавленным реактивом.
Бугер установил, что при прочих равных условиях слои вещества одинаковой толщины поглощают одну и ту же часть падающего на них светового потока. При этом поглощение светового потока не зависит от абсолютной интенсивности падающего света. Зависимость между интенсивностью монохроматического светового потока после прохождения его через раствор и интенсивностью падающего монохроматического светового потока выражается убывающей показательной функцией
 (1.14)
(1.14)
где Фо – интенсивность падающего монохроматического светового потока; Ф – интенсивность монохроматического светового потока после прохождения его через раствор; е – основание натуральных логарифмов; 1 – толщина слоя; k – коэффициент поглощения лучей, зависящий от природы вещества и длины волны светового потока.
| Темпера-тура°С | Поправки на температуру при содержании сухих веществ, % | ||||||||||||||
| От найденного содержания сухих веществ нужно отнять | |||||||||||||||
| 0,50 | 0,54 | 0,58 | 0,61 | 0,64 | 0,66 | 0,68 | 0,72 | 0,72 | 0,73 | 0,74 | 0,75 | 0,76 | 0,78 | 0,79 | |
| 0,46 | 0,49 | 0,53 | 0,55 | 0,58 | 0,60 | 0,62 | 0,64 | 0,65 | 0,66 | 0,67 | 0,68 | 0,69 | 0,70 | 0,71 | |
| 0,42 | 0,45 | 0,48 | 0,50 | 0,52 | 0,54 | 0,56 | 0,57 | 0,58 | 0,59 | 0,60 | 0,61 | 0,61 | 0,63 | 0,63 | |
| 0,37 | 0,40 | 0,42 | 0,44 | 0,46 | 0,46 | 0,48 | 0,49 | 0,50 | 0,51 | 0,52 | 0,53 | 0,54 | 0,54 | 0,55 | |
| 0,33 | 0,35 | 0,37 | 0,39 | 0,40 | 0,41 | 0,42 | 0,43 | 0,44 | 0,45 | 0,45 | 0,46 | 0,46 | 0,47 | 0,48 | |
| 0,27 | 0,29 | 0,31 | 0,33 | 0,34 | 0,34 | 0,35 | 0,36 | 0,37 | 0,37 | 0,38 | 0,39 | 0,39 | 0,40 | 0,40 | |
| 0,22 | 0,24 | 0,25 | 0,26 | 0,27 | 0,28 | 0,28 | 0,29 | 0,30 | 0,30 | 0,30 | 0,31 | 0,31 | 0,32 | 0,32 | |
| 0,17 | 0,18 | 0,19 | 0,20 | 0,21 | 0,21 | 0,21 | 0,22 | 0,22 | 0,23 | 0,23 | 0,23 | 0,23 | 0,24 | 0,24 | |
| 0,12 | 0,13 | 0,13 | 0,14 | 0,14 | 0,14 | 0,14 | 0,15 | 0,15 | 0,15 | 0,15 | 0,16 | 0,16 | 0,16 | 0,16 | |
| 0,06 | 0,06 | 0,06 | 0,07 | 0,07 | 0,07 | 0,07 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | |
| К найденному содержанию сухих веществ нужно прибавить | |||||||||||||||
| 0,06 | 0,07 | 0,07 | 0,07 | 0,07 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | 0,08 | |
| 0,13 | 0,13 | 0,14 | 0,14 | 0,15 | 0,15 | 0,15 | 0,15 | 0,15 | 0,16 | 0,16 | 0,16 | 0,16 | 0,16 | 0,16 | |
| 0,19 | 0,20 | 0,21 | 0,22 | 0,23 | 0,23 | 0,23 | 0,23 | 0,24 | 0,24 | 0,24 | 0,24 | 0,24 | 0,24 | 0,24 | |
| 0,26 | 0,27 | 0,28 | 0,29 | 0,30 | 0,30 | 0,31 | 0,31 | 0,31 | 0,31 | 0,31 | 0,32 | 0,32 | 0,32 | 0,32 | |
| 0,33 | 0,35 | 0,36 | 0,37 | 0,38 | 0,38 | 0,39 | 0,40 | 0,40 | 0,40 | 0,40 | 0,40 | 0,40 | 0,40 | 0,40 | |
| 0,40 | 0,42 | 0,43 | 0,44 | 0,45 | 0,46 | 0,47 | 0,48 | 0,48 | 0,48 | 0,48 | 0,48 | 0,48 | 0,48 | 0,48 | |
| 0,48 | 0,50 | 0,52 | 0,53 | 0,54 | 0,55 | 0,55 | 0,56 | 0,56 | 0,56 | 0,56 | 0,56 | 0,56 | 0,56 | 0,56 | |
| 0,56 | 0,57 | 0,60 | 0,61 | 0,62 | 0,63 | 0,64 | 0,64 | 0,64 | 0,64 | 0,64 | 0,64 | 0,64 | 0,64 | 0,64 | |
| 0,64 | 0,66 | 0,68 | 0,69 | 0,71 | 0,72 | 0,73 | 0,73 | 0,73 | 0,73 | 0,73 | 0,73 | 0,73 | 0,73 | 0,73 | |
| 0,72 | 0,74 | 0,77 | 0,78 | 0,79 | 0,80 | 0,80 | 0,81 | 0,81 | 0,81 | 0,81 | 0,81 | 0,81 | 0,81 | 0,81 |
При расчетах удобнее пользоваться десятичными логарифмами. Для перехода логарифмов от натуральных к десятичным применяют формулу
 (1.15)
(1.15)
где k – коэффициент погашения лучей (к = 2,3026 k').
При одной и той же длине волны светового потока коэффициенты поглощения и погашения зависят только от природы вещества. Коэффициент погашения равен обратной величине толщины слоя вещества, ослабляющего интенсивность проходящего через него светового потока в 10 раз, т. е.
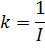
Беер установил, что между концентрацией растворенного окрашенного вещества и поглощающей способностью раствора имеется прямая пропорциональная зависимость

где ε – постоянная величина, зависящая от природы растворенного вещества и длины волны светового потока, но не зависящая от его концентрации; с – концентрация вещества в растворе.
Закон Бугера – Ламберта рассматривает изменения поглощения светового потока средой, пропускающей свет, при изменении толщины этой среды. Закон Беера устанавливает изменения поглощения светового потока одинаковой толщины при изменении концентрации вещества.
В отличие от закона Бугера – Ламберта закон Беера имеет много исключений, которые следует учитывать при фотоколориметрировании. В практике меняются не только концентрации, но и толщина слоя растворов.
Объединяя приведенные уравнения, получим выражение основного закона фотоколориметрии Бугера – Ламберта – Беера для растворов
 (1.17)
(1.17)
Выразив концентрацию растворенного вещества в грамм-молях на 1 л, а толщину слоя – в сантиметрах, получим коэффициент молярного погашения (ε').
При постоянной длине волны поглощаемого светового потока и одной и той же температуре коэффициент молярного погашения – величина постоянная для каждого вещества и в зависимости от строения вещества меняется в очень широких пределах. Так, для хромата калия ε'=500, а для роданида железа ε' = 100. Пользоваться уравнением (1.17) сложно, так как величины концентрации и толщины слоя находятся в показателе степени. Для приведения его к виду, удобному для расчета, и выведения некоторых фотометрических величин его преобразовывают в следующее уравнение:
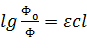 (1.18)
(1.18)
Левую часть уравнения (1.18), показывающую отношение интенсивности падающего светового потока к интенсивности светового потока, прошедшего через раствор, называют экcтинкцией, или погашением, и обозначают Е:
 (1.19)
(1.19)
Натуральный логарифм отношения интенсивности падающего светового потока к интенсивности прошедшего через раствор светового потока называют оптической плотностью, или поглощением, и обозначают D:
 (1.20)
(1.20)
Из сопоставления уравнений (1.19), (1.20) следует, что между оптической плотностью и экстинкцией имеется соотношение
E = 2,3026D (1.21)
Экстинкция и оптическая плотность пропорциональны концентрации вещества в растворе. Отношение интенсивности светового потока, прошедшего через раствор, к интенсивности падающего светового потока называют прозрачностью (пропусканием) вещества и обозначают Т:
 (1.22)
(1.22)
Величина Т, отнесенная к толщине слоя 1 см, называется коэффициентом светопропускания, который показывает, какая часть световой энергии проходит через испытуемый раствор.

Концентрация Концентрация
Рис. 5. Графическое выражение законов поглощения света:
а — зависимость пропускания (прозрачности раствора) от его концентрации; б — зависимость поглощения света от концентрации раствора.
Коэффициент пропускания меняется от 1 до 0 или от 100 % до 0.
Зависимость поглощения света или экстинкции от концентрации растворенного вещества (рис. 5) можно изобразить графически. Для этого на оси абсцисс откладывают концентрацию вещества, а на оси ординат – значение Т или экстинкции. В первом случае закон графически выражается кривой, во втором – прямой линией, наклон которой зависит от коэффициента молярного погашения. Обе линии проходят через начало осей координат.
Если испытуемый раствор подчиняется основному закону фотоколориметрии, то график, выражающий зависимость экстинкции или оптической плотности от концентрации раствора, будет представлен прямой линией. Если раствор не подчиняется этому закону, то прямолинейность на каком-либо участке или по всей линии нарушается. Поэтому перед проведением анализа выясняют, при каких концентрациях применим основной закон.
Практика показывает, что при высоких концентрациях растворов особенно часто нарушается основной закон фотоколориметрии. Причиной этого является взаимодействие молекул растворенного вещества друг с другом и с молекулами растворителя.
Основной закон фотоколориметрии нарушается не только при исследовании концентрированных растворов. Причины разнообразны: влияние электролитов, диссоциация веществ, изменение рН и др.

Рис. 6. Фотоэлектроколориметр ФЭК-56-М-У42:1 – рукоятка шторки; 2 – клемма для подсоединения к блоку питания; 3 – рукоятка переключения кювет: 4 – рукоятка правого барабана; 5 – индикатор; 6 – шкала левого барабана.
У одних окрашенных соединений окраска возникает постепенно и через некоторое время как бы «созревает», у других – образовавшаяся максимально интенсивная окраска постепенно бледнеет. Поэтому перед фотоколориметрированием устанавливают интервал времени, в котором окраска достигает максимальной величины и устойчива в течение времени, достаточного для колориметрирования. Например, для определения по Гриссу нитритов в мясных продуктах рекомендуется проводить колориметрирование через 15 мин после внесения реактивов в испытуемую вытяжку.
Образование окрашенного соединения, его интенсивность зависят и от других факторов: количества и концентрации применяемых реактивов, порядка их внесения, нестойкости окраски и ее изменения во времени, вызванного изменением химического состава растворенного вещества, образования даже небольшого количества других веществ, имеющих собственную окраску и меняющих оттенок исследуемого раствора.
При проведении фотоэлектроколориметрии не требуются стандартные растворы для сравнения с исследуемым образцом. Достаточно приготовить одну серию растворов с известной и различной концентрацией определяемого вещества, установить зависимость силы фототока от концентрации стандартных растворов и построить график этой зависимости. Пользуясь графиком зависимости силы тока от концентрации, определяют концентрацию испытуемого раствора.
Отечественная промышленность изготовляет различные фо-тоэлектроколориметры. Наиболее совершенными и универсальными являются фотоколориметры типа ФЭК, выпускаемые под марками ФЭК-56М-У42 (рис. 6), ФЭК-Н-57 и ФЭК-60.
В фотоэлектроколориметре ФЭК-Н-57 приемниками лучистой энергии служат два сурьмяно-цезиевых фотоэлемента (СЦВ-3) с внешним фотоэффектом. Фотоэлементы соединены со стрелочным нуль-гальванометром по дифференциальной схеме через усилитель. При измерении коэффициента пропускания абсолютная погрешность прибора не превышает 1 %. Прибор снабжен четырьмя одинаковыми наборами кювет с рабочим расстоянием от 1 до 50 мм. Источником света служат лампы накаливания СЦ-98 или СЦ-61.
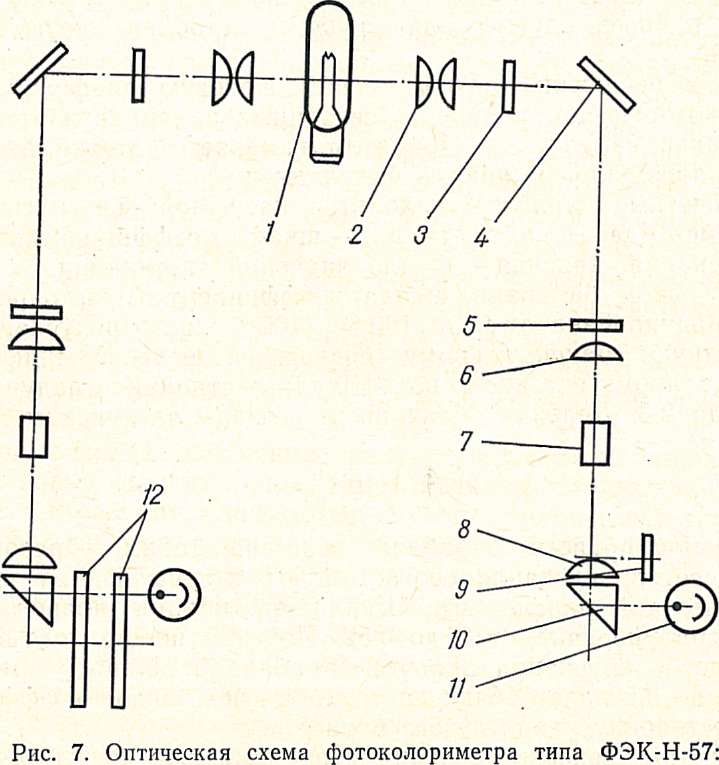
1 – осветительная лампа; 2 – конденсатор; 3 – теплозащитное стекло; 4 – зеркало;5 – светофильтр; 6 – линза; 7 – кювета; 8 – линза; 9 – измерительная щелевая диафрагма; 10 – призма; 11 – фотоэлемент; 12 – нейтральные клинья.
Фотоколориметр состоит из собственно прибора и питающего устройства, который предназначен для стабилизации напряжения осветительной лампы и электрической системы фотоэлементов. Оптическая схема фотоэлектроколориметра типа ФЭК изображена на рис. 7.Световой поток от лампы накаливания через конденсаторы и теплозащитные стекла, поглощающие тепловые лучи, попадает на отражающие зеркала. Отраженные световые потоки проходят через светофильтры, кюветы с раствором, линзы, призмы и попадают на фотоэлементы. При равенстве фототоков стрелка гальванометра останавливается на нуле. Левый световой поток дополнительно проходит через нейтральные клинья, которые его плавно ослабляют. Один из компенсирующих клиньев служит для грубой настройки, другой – для точной.
Световой поток проходит через щелевую диафрагму, ширина которой при вращении связанных с ней двух отсчетных барабанов изменяется. При этом изменяется также лучистый поток, падающий на правый фотоэлемент.
Отсчетные барабаны находятся на одной оси, каждый из них имеет две шкалы: черная – шкала коэффициентов светопропускания, красная – шкала значений экстинкции.
На левом барабане шкала коэффициентов светопропускания градуирована от 0 до 100% (100% светопропускания соответствуют максимальному раскрытию щелевой диафрагмы, а 0 – полному ее закрытию). Шкала экстинкции градуирована от 0 до 2,5. Деления обеих шкал связаны следующим соотношением:
 (1.23)
(1.23)
Шкала правого барабана нанесена таким образом, что 30 % светопропускания соответствуют максимальному раскрытию щелевой диафрагмы. Шкала экстинкции правого барабана градуирована от 0 до 0,52. Поэтому шкала коэффициентов светопропускания правого барабана в области измерений от 0,3 до 0,52 дает большую точность, чем шкала коэффициентов светопропускания левого барабана.
Чувствительность гальванометра изменяют регулятором на левой боковой стенке прибора. Рукоятка регулятора имеет три положения, соответствующие минимальной, средней и максимальной чувствительности. Для установки электрического нуля пользуются рукояткой на правой боковой стенке прибора, связанной с движком потенциометра.
Прибор снабжен двумя наборами светофильтров, которые вмонтированы в диски, связанные между собой тягами. Светофильтры переключают рукояткой на передней стенке прибора.
Питающее устройство состоит из стабилизатора напряжения, трансформатора и выпрямителя тока. С прибором оно соединено многожильным кабелем.
Порядок проведения анализа. В правый кюветодержатель помещают кювету с исследуемым раствором,в левый – с растворителем. При этомщелеваядиафрагма полностью открыта, а левый барабан установлен на100%-ноеделение почерной шкале. В результате поглощения света исследуемым раствором освещенность правого фотоэлемента будет меньше освещенности левого и стрелка гальванометра отклонится от нулевого положения. Для уравнивания фототоков в левый световой поток вводят нейтральный клин и ослабляют его до установки стрелки гальванометра на нуль. Затем в правом световом потоке кювету с исследуемым раствором заменяют кюветой с растворителем. При этом равенство фототоков нарушается. Вращением отсчетного барабана уменьшают ширину щели и ослабляют световой поток, падающий на правый фотоэлемент.
По шкале измерительного барабана отсчитывают коэффициент светопропускания, или экстинкцию исследуемого раствора. Наличие в приборе светофильтров позволяет значительно увеличить точность колориметрического анализа.
Светофильтры представляют собой окрашенные стекла, которые избирательно поглощают лучи определенной длины волны и поэтому кажутся окрашенными. Для увеличения чувствительности измерений светофильтр подбирают так, чтобы пропускаемый им свет поглощался анализируемым веществом. При этом все посторонние лучи, действующие на фотоэлементы, срезаются.
Перед началом измерений следует убедиться в правильности установки осветительной лампы. Для этого перед линзами у кювет помещают папиросную бумагу. Если осветительная лампа установлена правильно, то на папиросной бумаге наблюдается четкое изображение спиральной нити. До начала измерений нужно проверить нулевое положение стрелки гальванометра и электрического нуля. На нулевое положение гальванометр устанавливают рукояткой на наружной стенке прибора, а электрический нуль – после 20-минутной засветки фотоэлементов. В момент проверки световые пучки перекрывают шторкой, рукоятку чувствительности гальванометра устанавливают в положение «2» и, вращая рукоятку потенциометра, расположенную на правой стенке прибора, добиваются перемещения стрелки гальванометра в нулевое положение.
Измерения на приборе можно производить двумя способами.
Первый способ.Вправый пучок света помещают кювету с исследуемым раствором, в левый – кювету с растворителем, содержащим все вещества за исключением испытуемого. Левый барабан устанавливают на нулевое деление шкалы экстинкции. Ставят рукоятку чувствительности гальванометра в положение «1» и вращением рукоятки фотометрических клиньев стрелку гальванометра приводят к нулю. Нулевое положение стрелки гальванометра уточняют при большей чувствительности. Затем рукоятку чувствительности снова возвращают в положение «1» и, вращая кюветодержатель,в правый пучок света вводят кювету с растворителем. Вращением измерительных барабанов отклонившуюся стрелку гальванометра вновь устанавливают на нуль и уточняют нулевое положение стрелки при увеличенной чувствительности прибора. Экстинкцию отсчитывают по шкале левого барабана. Операцию повторяют три раза и вычисляют среднее значение экстинкции.
Второй способ. В оба пучка света помещают кюветы с растворителем. Правый барабан устанавливают на нулевое деление шкалы экстинкции. Вращением фотометрических клиньев устанавливают стрелку гальванометра на нуль. Затем в правый пучок света вводят кювету с исследуемым раствором. Вращением измерительных барабанов устанавливают стрелку гальванометра на нуль и производят отсчет по шкале правого барабана.
Построение калибровочных кривых. Чтобы получить данные для построения калибровочных кривых, готовят не менее 5-6 растворов с убывающей концентрацией, обращая внимание на точность измерения количества взятых веществ.
Окрашенные растворы последовательно фотометрируют, соблюдая одинаковое время от начала приготовления до измерения каждого стандартного раствора. Предварительно определяют рабочую зону, замеряя исследуемые растворы с минимальной и максимальной концентрацией. Концентрации стандартных растворов должны охватывать возможные изменения концентрации определяемого вещества в исследуемых растворах.
Экстинкцию находят как среднее значение из 3-5 определений.
Калибровочную кривую строят следующим образом. На лист миллиметровой бумаги размером 250X250 мм наносят две прямоугольные оси координат. На оси абсцисс откладывают по возрастающей концентрацию стандартных растворов, а на оси ординат – соответствующие экстинкции. Прямолинейный участок кривой на графике свидетельствует о подчинении стандартного раствора основному закону колориметрии. Если намечается изгиб линии, то необходимо нанести много точек, чтобы начало изгиба было зафиксировано с большей точностью. При массовых анализах калибровочные кривые можно заменить калибровочными таблицами, а при малом количестве измерений можно пользоваться расчетными формулами. Экстинкция находится в линейной зависимости от концентрации. Определив экстинкцию исследуемого вещества и зная ее величину для стандартного раствора, составляют пропорцию и вычисляют концентрацию вещества в исследуемом растворе.
Для выбора светофильтра ориентировочно можно пользоваться табл. 6.Чтобы более точно подобрать светофильтр, необходимо снять предварительно спектрофотометрическую кривую на приборе ФЭК-Н-57 или на спектрофотометрах СФ-14, СФ-16, СФ-26 и др.
Перед включением прибора рукоятку переключения чувствительности нужно установить в положение «0». Во избежание повреждения прибора включать его на более высокую чувствительность можно только после предварительной установки стрелки гальванометра на нулевое деление шкалы при меньшей чувствительности.
Таблица 6
| Окраска исследуемого вещества | Длина волны поглощаемого света, нм | Цвет требуемогосветофильтра | Длина волны пропускания света, нм | Номер светофильтров в фотоэлектро-колориметре ФЭК-Н-57 |
| Зеленовато-желтая | Фиолетовый.............. | 400—430 | 1.2 | |
| Желтая...................... | Синий, фиолетовый... | 420—450 | 1,2 | |
| Оранжевая............... | Синий......................... | 430—460 | ||
| Красная.................... | Зеленый...................... | 460—500 | ||
| Пурпурная............... | Зеленый...................... | 490—530 | ||
| Фиолетовая.............. | Зелено-желтый.......... | 520—550 | ||
| Красно-синяя........... | Желтый...................... | 520—550 | ||
| Сине-зеленая........... | Красный.................... | 600—630 | 7,8 |
ХРОМАТОГРАФИЯ
Хроматографические методы широко применяют при проведении исследований,не выполнимые другими инструментальными методами.Хроматографию как метод разделения впервые открыл в 1903 г. русский ученый М. С. Цвет, применив принцип адсорбции для анализа хлорофилла. В настоящее время хроматография охватывает различные методы разделения.
Хроматографические методы классифицируют в соответствии с выбранным типом подвижной и неподвижной фаз. В зависимости от агрегатного состояния фаз различают твердо-жидкостную (ТЖХ), жидко-жидкостную (ЖЖХ), газоадсорбиционную (ГАХ), газожидкостную (ГЖХ) хроматографию, а в зависимости от природы механизма разделения – распределительную, адсорбционную, ионообменную и др.
В распределительной хроматографии вещества распределяются между двумя жидкими фазами, одна из которых неподвижная.
Адсорбционная хроматография основана на сорбции растворенного вещества поверхностью твердой фазы,
ионообменная – на образовании ионных Соединений между растворенными веществами и заряженными группами сорбента.
По виду вспомогательных средств различают тонкослойную, бумажную, газовую, газожидкостную и колоночную хроматографию.
Тонкослойная хроматография
Тонкослойная хроматография – очень удобный метод разделения небольших количеств многокомпонентных смесей веществ. Преимущества этого метода перед бумажной хроматографией: быстрота разделения веществ, устойчивость слоя к агрессивным проявителям и нагреванию и часто значительно большаячувствительность, чем на бумаге, что позволяет обнаружить в случае применения некоторых реагентов ничтожно малые количества разделяемых веществ (от 0,1 до 0,005 мкг).
При использовании тонкослойной хроматографии для количественного определения применяют тонкий слой силикагеля, нанесенный на стандартные стеклянные пластинки; для осуществления качественного анализа веществ – слайды (слой силикагеля, нанесенный на предметное стекло), а также силуфольные пластинки, содержащие флуоресцентный индикатор.
В зависимости от того, в каком направлении поступает растворитель на пластинку, различают методы восходящей, нисходящей и горизонтальной хроматографии.
Для разделения веществ методом тонкослойной хроматографии применяют как одномерную, так и двумерную хроматографию.
Для тонкослойной хроматографии необходим сорбент, который не содержит примесей тяжелых металлов и органических веществ. Примеси железа, присутствуя, в частности, в силикагеле, катализируют окисление жирных кислот, вызывают изомеризацию органических соединений.
Силикагель, применяемый в тонкослойной хроматографии, должен иметь крупнопористое строение, размер зерна – 150-200 меш (размер отверстия сита – соответственно 0,10-0,07 мм). Для тонкослойной хроматографии чаще всего используют готовый силикагель.
За рубежом некоторые фирмы («Дезага», «Гейдельберг», «Мерк», «Дармштадт» и др.) готовят специально силикагель G, кизельгур С и окись алюминия Gс добавкой 5 % гипса и без добавки его.
Чтобы получить закрепленный слой, сорбент закрепляют на стеклянной пластине фиксатором: гипсом медицинским, штукатурным, рисовым или маисовым крахмалом.
Пластинки с закрепленным слоем сорбента готовят следующим образом: на воздухе при комнатной температуре в течение 15-20 мин на горизонтальной поверхности моют и сушат стеклянные пластинки подходящего размера (20X15, 20Х10, 20x20 см), устанавливают прибор для нанесения слоя, готовят сорбционную массу в виде суспензии из сорбента, фиксатора и воды и аппликатором разравнивают ее по поверхности стеклянных пластинок. После сушки пластинки активизируют нагреванием в сушильном шкафу или выдерживанием на воздухе.
Для пяти пластинок размером 20X20 см с толщиной слоя сорбента 250 мкм берут 25-30 г силикагеля, содержащего 5 % гипса, смешивают в ступке с 35 мл воды, Затем к полученной пасте при перемешивании добавляют еще 15 мл воды. Суммарная продолжительность перемешивания не должна превышать 100 с.
Полученную суспензию выливают в аппликатор, посредством которого ее переносят на пластинку.
Суспензию можно приготовить и другим способом. Берут 25 г сорбента, прибавляют 50 мл дистиллированной воды, все тщательно перемешивают в колбе с пробкой и выливают в аппликатор. Чтобы устранить побочный эффект, пластинки освобождают от сорбента по периметру на 5-7 мм.
Готовые пластинки хранят в эксикаторе, содержащем влагопоглотитель: силикагель,H2SO4,СаС12.
Пробы испытуемых веществ (от 5 до 1000 мкг) наносят на расстоянии 1,5 см от нижнего края пластинки сплошной полосой (капля к капле) или каплей. Для нанесения пробы применяют микропипетки или откалиброванные микрошприцы. При этом слой сорбента не должен нарушаться.
Ниже рассмотрено разделение веществ методом восходящей хроматографии в закрепленном слое. Камера для хроматографирования на закрепленном слое сорбента представляет собой подходящий по размеру пластинки стеклянный сосуд любой формы с плоским дном. Высота камеры должна быть примерно 25 см при размере пластинки 20X20 см.
В сосуд наливают растворитель или систему растворителей в таком количестве, чтобы поставленная вертикально пластинка с нанесенным веществом погружалась в растворитель на 5 мм ниже вещества, нанесенного для разделения.
Для насыщения камеры парами растворителей к задней стенке ее прикрепляют смоченный растворителем лист фильтровальной бумаги, достигающий дна сосуда. Сверху камеру закрывают пришлифованной крышкой или стеклом. Высота подъема жидкости по слою сорбента не должна превышать 10-11 см, так как при большем пробеге растворителя наблюдаются сильное замедление продвижения жидкости и диффузия пятен.
После того, как жидкость поднимется на указанную высоту, пластинку вынимают и отмечают линию фронта, а также высоту подъема растворителя. Затем пластинку сушат в вытяжном шкафу в токе воздуха.Для разделения веществ обычно растворитель пропускают по пластинке с сорбентом один раз. Если в одной системе растворителей не все вещества смеси разделяются одинаково хорошо, то применяют так называемое ступенчатое, или повторное, хроматографирование в разных системах растворителей.
Сначала применяют систему, которая делит менее полярные вещества. При этом растворитель продвигается вперед почти до конца пластинки. Затем хроматограмму сушат для удаления растворителя, и хроматографирование проводят в другой системе растворителей, которую пропускают на более короткое расстояние, чем первую.
Чтобы обнаружить вещества, поглощающие ультрафиолетовые лучи в определенной области спектра, часто применяют слои сорбента, содержащие флуоресцирующее вещество, либо опрыскивают хроматограмму раствором флуоресцирующего вещества или веществ, поглощающих ультрафиолетовые лучи в определенной области спектра, или пользуются источниками света с максимумами излучения в области 254 и 365 нм. Для этого применяют следующие приборы: ультрахимескоп, дающий максимум излучения в области 254 нм, и настольный ультрафиолетовый осветитель с максимальным излучением около 365 нм.
Если оптическими методами нельзя обнаружить вещества на хроматограммах, то применяют химические методы проявления.
Для проявления парами йода пластинку помещают в эксикатор с кристаллами йода в разогретой фарфоровой чашке. Через 10-15 мин пластинку вынимают и оставляют на воздухе до испарения избытка йода. При этом на светлом фоне образуются окрашенные пятна веществ.
Метод рекомендуется для определения непредельных и других соединений. Для проявления отдельных пятен наиболее широко применяют опрыскивание пластинки реактивами, дающими цветные реакции с разделенными веществами. Опрыскивание проводят в стеклянном колпаке.
При идентификации веществ определяют пробег исследуемого вещества (фракции) от линии старта. Это определение не является достаточным, так как Rfхарактеризует лишь относительное положение пятна внутри хроматограммы.
Для точного указания условий анализа пользуются величиной Rst, определяемой как отношение расстояния исследуемого вещества от старта к расстояний пятна от старта.
Количество вещества в пятне определяют по площади его поверхности, которая зависит от количества нанесенного вещества, толщины и активности слоя, а также объема нанесенного раствора.
Пробы анализируемых веществ на одной пластинке сравнивают с пробой эталонных образцов на другой. Для этого измеряют площади пятен. На основании количества взятого вещества и найденной площади строят калибровочную кривую.
Для количественного определения разделенных веществ применяют также фотометрические методы.
Анализ природных липидов. Методом восходящей хроматографии в закрепленном слое можно получить общее представление о составе исследуемой смеси липидов. Этим методом липиды и продукты гидролиза хорошо разделяются на отдельные классы соединений. Перед разделением липидов из продуктов животного происхождения их извлекают методом экстракции, соблюдая следующие основные правила:
все операции (гомогенизацию, экстракцию и др.) проводить в атмосфере азота, чтобы избежать автоокисления ненасыщенных липидов. Для этого можно применять аппарат Сокслета или экстрактор и использовать только очищенный и свежеприготовленный растворитель;
придерживаться соотношения массы, полученной при измельчении продукта, и экстрагирующего раствора (не менее 1:25);
нагревать растворы, содержащие липиды, только в случае крайней необходимости;
удалять из липидных экстрактов все нелипидные составные части без потерь липидов;
хранить экстрагированный материал в условиях, обеспечивающих минимальное изменение липида: в петролейном эфире, хлороформе, в атмосфере азота, в холодильнике, в запаянных ампулах.
В зависимости от фракционируемого класса липидов применяют следующие растворители в соотношении по объему: жирные кислоты – растворители; ледяная уксусная кислота – вода (24:1); жирные кислоты и их метиловые эфиры – растворители: ледяная уксусная кислота – вода (3:1); холестериновые эфиры высших жирных кислот – растворители: метилэтилкетон – ацетонитрил (7:3); диглицериды – растворители: хлороформ – метанол – вода (5:15:1).
Для разделения липидов на закрепленном силикагеле применяют растворители в различных соотношениях: петролейный эфир-диэтиловый эфир в соотношении 80:20 по объему – для альдегидов и моноглицеридов; петролейный эфир – диэтиловый эфир – ледяная уксусная кислота (80:20:1) – для смеси липидов, содержащих свободные жирные кислоты; гексан – эфир – уксусная кислота (90:10:1) – для нейтральных липидов; хлороформ – метанол – вода (65:25:4) – для фосфолипидов.
При изучении фосфолипидных фракций, входящих в состав желтка куриных яиц, используют адсорбционную хроматографию в тонком (0,5 мм) незакрепленном слое силикагеля КСК отечественного производства. Размолотый, отмученный и очищенный от посторонних примесей силикагель с частицами размером 30-50 мкм (200 меш) наносят посредством аппликатора и специального устройства на стеклянную пластинку размером 10X20 см.
Перед использованием готовой пластинки нанесенный слой промывают метанолом, чтобы освободить рабочий слой силикагеля от возможных органических примесей.
Для разделения фосфолипидов непосредственному фракционированию подвергают общие липиды, растворенные в хлороформе, которые в количестве 30 мкг наносят по фосфору тонкой полоской длиной 5 см на пластинку.
Чтобы получить отдельные фракции фосфолипидов, липиды разгоняют в нейтральной системе хлороформ – метанол – вода (65:25:4). Для этого используют высокие стеклянные камеры с пришлифованными крышками, которые устанавливают на строго горизонтальную поверхность.
Чтобы выявить расположение отдельных фракций фосфолипидов, пластинки после просушивания под тягой помещают в эксикатор с парами йода либо опрыскивают 0,04 %-ным спиртовым раствором 2,7-дихлорфлюоросцеина и просматривают в ультрафиолетовом свете. В первом случае на пластинках в местах расположения фосфолипидов на белом фоне видны желто-коричневые полосы, во втором – на светло-сером фоне наблюдается желтое и фиолетовое свечение.
Расположение отдельных фракций фосфолипидов на тонком слое силикагеля от линии старта следующее: лизолецитины, сфингомиелины, серинфосфатиды, лецитины (холинфосфатиды), этаноламинфосфатиды, цереброзиды, прочие липиды.
Фосфолипиды идентифицируют по тестам – цветным реакциям. Для обнаружения фосфолипидов, содержащих аминогруппы (этаноламинфосфатиды и серинфосфатиды), пластинки обрабатывают нингидрином и наблюдают красное окрашивание пятен на белом фоне.
Холинсодержащие фосфолипиды (лецитины, лизолецитины, сфингомиелины) обнаруживают опрыскиванием пластинок реактивом Драгендорфа (1,7 г Bi(NO3)2·5Н2O в 20 %-ной уксусной кислоте +10 %-ный водный раствор KJ). В этом случае на пластинке появляются желто-оранжевые пятна на бледно-желтом фоне. Цереброзиды обнаруживают путем обработки хроматограммы раствором молибденовокислого аммония в смеси с соляной и хлорной кислотами
3 г(NH4)2МО4 + 25 мл Н2O+3мл НСl+15 мл 60%-ной НСlO4.
После нагревания пластин в течение 20 мин в сушильном шкафу при 110 °С пятно цереброзидов приобретает фиолетовую окраску, остальные пятна фосфолипидов – коричневую.
Для количественного определения отдельных липидных фракций отдельные пятна соскабливают с пластинки и переносят в пробирку объемом 20 мл, заливают три раза по 10 мл метанолом, взмучивают и настаивают каждый раз по 5 мин. Затем для лучшей очистки от силикагеля элюаты центрифугируют при частоте вращения 6000 об/мин и декантируют в мерные цилиндры со шлифом. Часть элюата (например, половину объема) используют для количественного определения фосфо-липидов.
О содержании определенного фосфолипида в липидах животной ткани судят по молярному содержанию фосфора в элюате. Для этого элюаты минерализуют в присутствии молибденовокислого аммония и аскорбиновой кислоты.
Окрашенные растворы фосфора после инкубирования коло-риметрируют на спектрофотометре (λ = 820 нм).
Для построений калибровочной кривой, необходимой при определении фосфора в элюате лецитина из пятна, применяют однозамещенный фосфорнокислый калий КН2РО4.
Анализ карбонильных соединений. Алифатические альдегиды и кетоны удобнее разделять в виде их 2,4-динитрофенилгидразонов (2,4-ДНФГ). Лучшей системой для разделения н-алифатических альдегидов является тонкий слой силикагеля – крахмал в бензоле, насыщенном водой (98:8,2 по объему).
2,4-динитрофенилгидразона тех же альдегидов, а также циси трансизомеры фурфуриловых альдегидов, бензальдегида и кетонов, глиоксаля и другие соединения идентифицируют на незакрепленном слое окиси алюминия в системе бензол – гексан (1:1) ив эфире.
Пятна на пластинке обнаруживают фосфорно-молибденовой кислотой.
На слое силикагель – гипс в системе гексан – этилацетат (7:3 по объему) разделяют коричный и бензойный альдегиды в виде 2,4-динитрофенилгидразонов в системе бензол – петролейный эфир (3:1 по объему).
Ряд моно-, ди- и триалкилциклогексанонов с алифатическими и териеновыми заместителями разделяют на закрепленном слое окиси алюминия в системе бензол-петролейный эфир при температуре кипения 40-60 °С (1:3 по объему). Пятна обнаруживают парами йода или раствором 2,4-динитрофенил-гидразона.
Анализ углеводов. Для разделения таких гидрофильных соединений, как углеводы, применяют модифицированные слои: кизельгур – гипс; силикагель – гипс; целлюлоза – гипс.
Смесьcахаров разделяют на слое кизельгур – гипс, пропитанном 0,02 М раствором ацетата натрия. Разделение Сахаров возможно также на основе силикагель – гипс, пропитанной борной кислотой. Для приготовления слоя к 4 г силикагеля добавляют 6 мл 0,1 н. раствора борной кислоты. В этом случае хроматографирование ведут в кислых системах.
Системы растворителей для Сахаров: 65 мл этилацетата + 35 мл раствора изопропанол – вода (2:1); бензол – метанол – уксусная кислота (1:3:1); метилэтилкетон – метанол – уксусная кислота (3:1:1); этилацетат – пиридин – вода (2:1:2); хлороформ – метанол (19:2, 19:3, 19:5).
При количественном анализе Сахаров их элюируют из пятен, выскобленных с пластинки, окисляют 0,05 н. или 0,01 и. раствором бихромата калия в 70%-ном растворе серной кислоты. Неиспользованный бихромат подвергают обратному титрованию.
Не нашли, что искали? Воспользуйтесь поиском: